
1,1-二取代-1,2-丙二烯化合物的合成
2012-12-23 03:08胡麟峰章彦婧毛国梁郑卫新
杭州师范大学学报(自然科学版) 2012年4期
胡麟峰,王 萍,章彦婧,洪 雅,陈 聪,毛国梁,郑卫新
(1.杭州师范大学材料与化学化工学院,浙江 杭州310036;2.东北石油大学化学化工学院,黑龙江 大庆 163318)
1,1-二取代-1,2-丙二烯化合物的合成
胡麟峰1,王 萍1,章彦婧1,洪 雅1,陈 聪1,毛国梁2,郑卫新1
(1.杭州师范大学材料与化学化工学院,浙江 杭州310036;2.东北石油大学化学化工学院,黑龙江 大庆 163318)
以炔烃为原料,经磺酸炔丙酯与格氏试剂的SN2’反应合成1,1-二取代-1,2-丙二烯型化合物.该方法具有原料简单易得、反应条件温和、副反应较少、产率高等优点.产物经1H NMR、13C NMR、IR以及MS确证.
1,1-二取代-1,2-丙二烯;磺酸炔丙酯;格氏试剂;SN2’反应;合成
1,2-丙二烯化合物是一类具有2个π-轨道同处于一个碳原子上结构的化合物,当其末端的4个取代基两两不同时,该分子就具有轴手性,在反应中该轴手性可以转移产生新的手性中心,为光学活性化合物的合成提供了一条途径.经过近20年的研究与发展,该类化合物在有机合成中可作为一类高效的“三碳原子结构”单元不饱和合成砌块,用于合成天然产物、医药化学以及材料科学中的目标分子[1-3].因此,合成1,2-丙二烯化合物具有重要的理论意义及应用价值.
根据底物不同,合成丙二烯化合物的方法可以分为以下几种类型:①从卤代烯烃衍生物出发,通过1,2-消除反应[4]、分子内 H 迁移-消除反应[5]、Wittig反应[6]、C—C键消除反应[7]、2-卤代-1,3-丁二烯类化合物烯丙位取代反应[8]合成丙二烯化合物;②从炔烃衍生物出发,通过炔烃在碱性条件的异构化反应[9]、SN2’反应[10]、炔丙基或联烯基金属中间体对羰基化合物的加成反应[11]、端炔的Crabbé反应[12]、钯催化交叉偶联反应[13]、经过锆杂中间体的反应[14]合成丙二烯化合物;③共轭烯炔衍生物通过1,4-加成反应[15]、烯炔的环金属化反应[16]以及含有杂原子的烯炔化合物的碳骨架重排反应[17]合成丙二烯化合物;④从环丙烷及其衍生物出发,通过重排反应[18-19]合成丙二烯化合物.其中炔丙位带有离去基团炔烃的SN2’反应可在形成1,2-丙二烯骨架的同时在双键碳原子上引入新基团,是合成多取代丙二烯的重要方法.然而炔丙位上的SN2竞争反应是该方法使用所要解决的主要问题.本文以炔烃为原料,经磺酸炔丙酯与格氏试剂的SN2’反应合成系列1,1-二取代1,2-丙二烯化合物的改进合成方法,该方法具有原料简单易得、反应条件温和、副反应较少、产率高等优点.
1 结果与讨论
实验中,原位生成的活性炔基锂首先与多聚甲醛反应生成炔丙醇衍生物1,继而经磺酰化后形成所需要的原料炔丙酯化合物2(图1).
室温下,以四氢呋喃为溶剂,磺酸炔丙酯2与格氏试剂在亚铜盐催化下经SN2’反应生成1,1-二取代丙二烯化合物4.反应如图2所示.
为减少炔丙位上直接取代的副产物4a’的生成,根据文献[8]方法,我们以2a为底物与2当量EtMg-Br的反应为模板,首先探索了催化剂对SN2’与SN2反应竞争的影响.结果如表1所示.
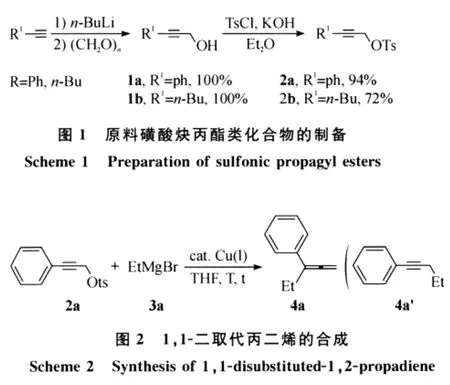
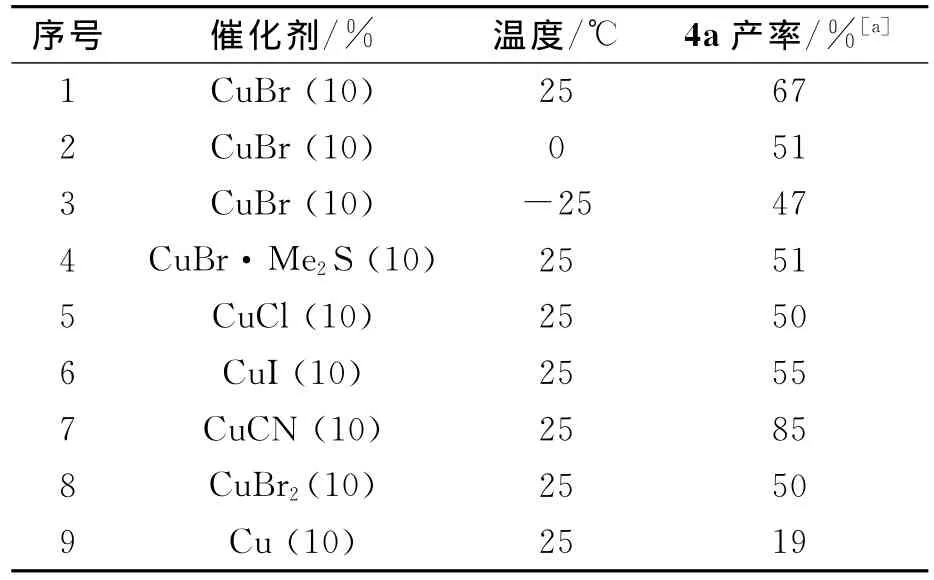
表1 催化剂对SN2’与SN2反应竞争的影响Tab.1 Influence of the catalysts on the competition between SN2’and SN2
从表1可知,反应温度在25℃,10%CuCN催化该反应可获得较高的产率(序号7).在此基础上,保持25℃的反应温度,我们对试剂用量进行探索.结果如表2所示.
综合表1(序号7)与表2(序号2)的结果,该模板反应的最优条件为25℃时1当量磺酸炔丙酯与1.5当量的格氏试剂在10%的CuCN催化下作用.与文献[8]方法相比,该反应减少了格氏试剂的用量,并选用CuCN作为催化剂获得了更高的产率.在此条件下,我们合成了系列1,1-二取代-1,2-丙二烯化合物(表3).
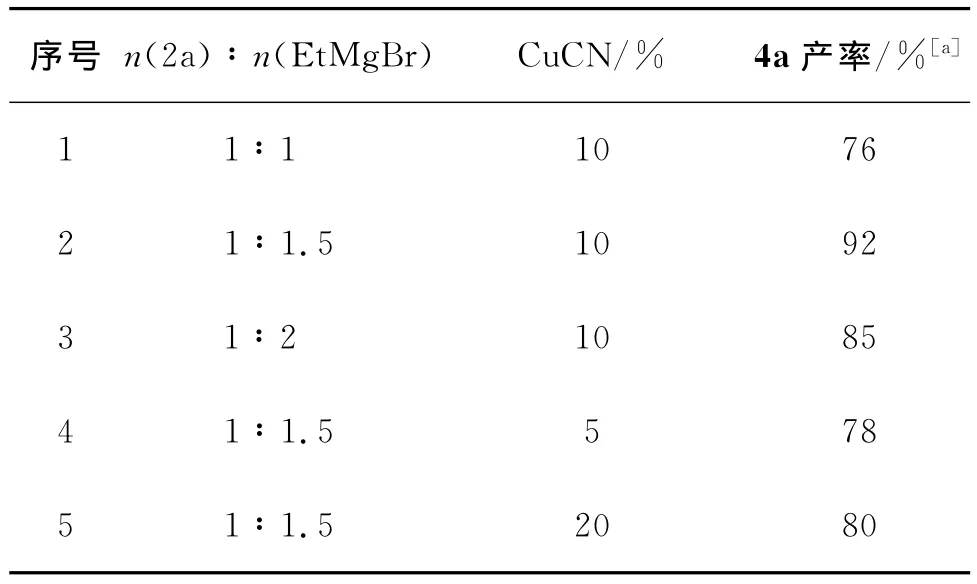
表2 试剂用量对反应的影响Tab.2 Influence of the ratio of the reagents
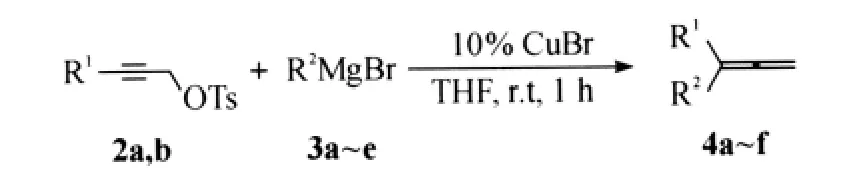
表3 系列1,1-二取代-1,2-丙二烯化合物的合成Tab.3 Synthesis of 1,1-disubstituted-1,2-propadienes
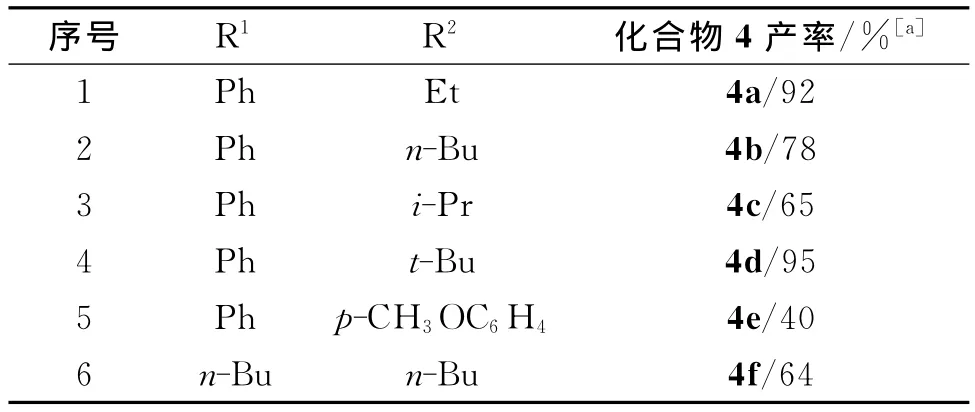
[a]分离产率,以炔丙酯2a为基准.
表3中为各种1,1-二取代丙二烯化合物的合成,产率中等至优良.化合物4a~f的IR光谱图中,1 950cm-1附近具有中等强度的吸收峰,是丙二烯累积双键骨架的特征伸缩振动峰,在1H NMR谱图中,1,1-二取代丙二烯化合物端位上2个氢的位移在δ5.0ppm附近,在13C NMR谱图中,丙二烯累积双键中间sp杂化的碳原子的化学位移在δ208ppm附近.
2 结 论
综上所述,通过磺酸炔丙酯与格氏试剂的SN2’反应可高产率地合成1,1-二取代-1,2-丙二烯化合物.该方法具有原料简单易得、反应条件温和、副反应较少、产率高等优点.
3 实验部分
温度计未校正.所有反应均在氮气氛围下进行,采用Schlenk技术操作,所有实验操作中用到的玻璃仪器、注射器、穿刺针均在120℃下干燥处理.柱层析分离采用300~400目硅胶.1H NMR采用Bruker AC2P400(400MHz)核磁共振谱仪测定,以TMS为内标,CDCl3作溶剂;红外光谱采用德国BrukerTENSO27原位红外仪,质谱采用Bruker Daltonics Esquire 3000plus测定.如非特别注明,所用原料均为购置且未经过重新处理.溶剂四氢呋喃(THF)在氮气氛围下使用钠/二苯甲酮处理重蒸.
3.1 炔丙醇 (1a,b)的制备
在氮气保护下,在100mL具塞圆底反应瓶中加入40mmol炔烃的40mL四氢呋喃中,冷却至-78℃,在搅拌下缓慢加入44mmol正丁基锂 (1.6M),滴加完毕后,加入40mmol多聚甲醛,将反应瓶转移至室温继续反应至完成.用饱和NaCl水溶液淬灭,分液,水相用乙醚萃取3次,合并有机相,无水MgSO4干燥,有机相经过滤、浓缩、柱层析分离得到产物1a,b.化合物表征如下:
1a[20]:3-苯基-2-丙炔-1-醇
浅黄色液体,Yield:100%;IR(cm-1)ν3 345,2 924,2 237,1 598,1 489,756,691,572;
1H NMR (CDCl3,Me4Si)δ(ppm):2.53(s,1H),4.48~4.50(d,J=6.0Hz,2H),7.24~7.44(m,5H);
13C NMR (CDCl3,Me4Si)δ(ppm):51.4,85.4,87.1,122.4,128.2,128.4,131.5;
MS(EI)m/z(%):132(M+,68),131(100).
1b[21]:2-己炔-1-醇
无色液体,Yield:100%;IR(cm-1)ν3 346,2 933,2 224,1 458,1 137,1 012,726;
1H NMR(CDCl3,Me4Si)δ(ppm):0.89~0.92(t,J =7.2Hz,3H),1.36~1.52(m,4H),2.20~2.23(t,J =6.8Hz,2H),2.81(s,1H),4.24(s,2H);
13C NMR (CDCl3,Me4Si)δ(ppm):13.4,18.2,21.7,30.5,50.9,78.1,86.1;MS(EI)m/z(%):112(M+,4),83(100).
3.2 磺酸炔丙酯化合物(2a,b)的制备
在250mL三颈瓶中加入24mmol对甲苯磺酰氯,3.36g氢氧化钾,溶于40mL无水乙醚中,在冰浴中剧烈搅拌下滴加20mmol炔丙醇1,滴加完毕后,继续搅拌至反应完全.用饱和NaCl水溶液淬灭,分液,水相用乙醚萃取3次,合并有机相,用无水MgSO4干燥,经过滤、浓缩、柱层析分离得到产物2.化合物表征如下:
2a[22]:对甲基苯磺酸-3-苯基2-丙炔-1-酯
白色针状晶体,Yield:94%;m.p.:81~82℃.IR(cm-1)ν2 950,1 347,1 294,1 189,1 174,821,695;
1H NMR(CDCl3,Me4Si)δ(ppm):2.39(s,3H),4.95(s,2H),7.26~7.86(m,9H);
13C NMR(CDCl3,Me4Si)δ(ppm):21.2,58.4,80.3,88.7,121.1,127.8,128.0,128.8,129.6,131.4,133.0,144.8;
MS(EI)m/z(%):286(M+,1),115(100).
2b[23]:对甲基苯磺酸-2-己炔-1-酯
无色液体,Yield:72%;IR(cm-1)ν2 959,2 873,2 310,2 237,1 598,1 369,1 170,1 096,939,664,555;
1H NMR(CDCl3,Me4Si)δ(ppm):0.84~0.88(t,J =7.2Hz,3H),1.25~1.40(m,4H),2.04~2.09(m,2H),2.44(s,3H),4.69~4.70(t,J =2.0Hz,2H),7.26~7.82(m,4H);
13C NMR(CDCl3,Me4Si)δ (ppm):13.4,18.2,21.5,21.7,30.0,58.7,71.7,90.5,128.0,129.6,133.3,144.7;
MS(EI)m/z(%):173(M+-C6H5CH3,6),91(100).
3.3 1,2-丙二烯化合物(4a~f)的制备
在氮气保护下,加入10mmol镁屑,一小粒碘,加入10mL四氢呋喃,缓慢滴加10mmol卤代烃,使得溶液保持微沸状态直至镁屑完全反应,待用.加入5mmol磺酸炔丙酯化合物2,10%氰化亚铜,溶于10 mL四氢呋喃中,冷却至0℃,在搅拌条件下缓慢滴加已制备好的格氏试剂,滴毕后缓慢升至室温,继续反应至完全.用饱和NH4Cl水溶液淬灭,分液,有机相用饱和NaCl水溶液洗涤,水相用乙醚萃取3次,合并有机相,无水MgSO4干燥,经过滤、浓缩、柱层析分离得到产物4a~f.化合物表征如下:
4a[25]:3-苯基-1,2-戊二烯
无色液体,yield:56%;IR(cm-1)ν3 060,1 940,1 493,1 220,696;
1H NMR(CDCl3,Me4Si)δ(ppm):1.13~1.17(t,J =7.6Hz,3H),2.41~2.43(m,2H),5.09(s,2H),7.16~7.41(m,5H);
13C NMR(CDCl3,Me4Si)δ(ppm):12.4,22.3,78.7,106.6,125.8,126.5,128.3,136.5,208.3;
MS(EI)m/z(%):144(M+,87),129(100).
4b[25]:3-苯基-1,2-庚二烯
无色液体,yield:48%;IR(cm-1)ν3 060,1 940,1 493,1 208,695;
1H NMR(CDCl3,Me4Si)δ(ppm):0.91~0.95(t,J =7.2Hz,3H),1.36~1.45(m,2H),1.50~1.57(m,2H),2.38~2.44(m,2H),5.04~5.06(t,J =3.6Hz,2H),7.16~7.41(m,5H);
13C NMR (CDCl3,Me4Si)δ(ppm):13.9,22.4,29.1,30.0,77.9,104.9,125.9,126.4,128.3,136.5,208.6;
MS(EI)m/z(%):172(M+,10),130(100).
4c[23]:4-甲基-3-苯基-1,2-戊二烯
无色液体,yield:55%;IR(cm-1)ν3 060,1 941,1 494,1 223,695;
1H NMR (CDCl3,Me4Si)δ(ppm)1.12~1.14(d,J =6.4Hz,6H),2.76~2.82(m,1H),5.06~5.07(d,J =2.8Hz,2H),7.15~7.40(m,5H);
13C NMR(CDCl3,Me4Si)δ(ppm)22.1,27.2,79.0,112.1,125.9,126.4,128.3,136.4,207.5;
MS(EI)m/z(%):158(M+,65),143(100).
4d[24]:4,4-二甲基-3-苯基-1,2-戊二烯
无色液体,yield:89%;IR(cm-1)ν3 056,1 949,1 490,1 244,700;
1H NMR(CDCl3,Me4Si)δ(ppm)1.13~1.14(t,J=4.0Hz,9H),4.75~4.77(t,J =3.6Hz,2H),7.19~7.27(m,5H);
13C NMR(CDCl3,Me4Si)δ(ppm)29.8,33.5,75.3,114.6,126.6,127.7,129.3,137.4,206.0;
MS(EI)m/z(%):172(M+,22),57(100).
4e[25]:1-苯基-1-(4’-甲氧基)苯基-1,2-丙二烯
浅黄色液体,yield:10%;IR(cm-1)ν3 059,2 925,1 943,1 599,1 495,1 452,1 040,755,692;1H NMR(CDCl3,Me4Si)δ(ppm)3.78(s,3H),5.22(s,2H),6.86~7.36(m,9H);
13C NMR(CDCl3,Me4Si)δ(ppm)55.2,77.8,108.6,113.8,127.1,127.2,128.2,128.3,129.5,136.5,158.8,209.6;
MS(EI)m/z(%):222(M+,100).
4f[25]:3-正丁基-1,2-庚二烯
浅黄色液体,yield:53%;IR (cm-1)ν3 047,2 958,1 957,1 465,843;
1H NMR(CDCl3,Me4Si)δ(ppm)0.88~0.91(t,J =7.2Hz,6H),1.28~1.44(m,8H),1.89~1.95(m,4H),4.60~4.64(m,2H);
13C NMR (CDCl3,Me4Si)δ(ppm)13.9,22.4,29.7,31.8,75.0,103.2,205.7;
MS(EI)m/z(%):123(M+-CH2CH3,2),68(100).
[1]Schuster H F,Coppola G M.Allenes in organic synthesis[M].New York:John Wiley &Sons,1984:1-385.
[2]Ma Shengming.Some typical advances in the synthetic applications of allenes[J].Chem Rev,2005,105(7):2829-2871.
[3]Pinho e Melo T M V D.Allenes as building blocks in heterocyclic chemistry[J].Monatsh Chem,2011,142(7):681-697.
[4]Kilbas B,Azizoglu A,Balci M.Incorporation of an allene unit intoα-pinene viaβ-elimination[J].Helv Chim Acta,2006,89(7):1449-1456.
[5]Tang Meng,Fan Chun'an,Zhang Fumin,et al.New metal-free one-pot synthesis of substituted allenes from enones[J].Org Lett,2008,10(24):5585-5588.
[6]Zhou Hongwei,Liu Guoliang,Zeng Changying.Bismetalated carbon for tandem Wittig-type reaction via allylgallation of magnesium acetylides:A convenient and efficient method to allyl allenes[J].J Organomet Chem,2008,693(4):787-791.
[7]Kolakowski R V,Manpadi M,Zhang Yue,et al.Allene synthesis via C-C fragmentation:method and mechanistic insight[J].J Am Chem Soc,2009,131(36),12910-12911.
[8]Ogasawara M,Ge Yonghui,Uetake K.Preparation of multisubstituted allenes from allylsilanes[J].J Org Chem,2005,70(10):3871-3876.
[9]Brossat M,Heck M P,Mioskowski C.Bistrimethylsilylpropargylic ether:A versatile ambident synthon to access substituted allenyne ethers andα-substituted bispropargylic alcohols[J].J Org Chem,2007,72(15):5938-5941.
[10]Pu Xiaotao,Ready J M.Direct and stereospecific synthesis of allenes via reduction of propargylic alcohols with Cp2Zr(H)Cl[J].J Am Chem Soc,2008,130(33):10874-10875.
[11]Park J,Kim S H,Lee P H.Selective indium-mediated 1,2,4-pentatrien-3-ylation of carbonyl compounds for the efficient synthesis of vinyl allenols[J].Org Lett,2008,10(21):5067-5070.
[12]Kuang Jinqiang,Ma Shengming.One-pot synthesis of 1,3-disubstituted allenes from 1-alkynes,aldehydes,and morpholine[J].J Am Chem Soc,2010,132(6):1786-1787.
[13]Ohmiya H,Yang Mingyu,Yamauchi Y,et al.Selective synthesis of allenes and alkynes through ligand-controlled,palladium-catalyzed decarboxylative hydrogenolysis of propargylic formates[J].Org Lett,2010,12(8):1796-1799.
[14]Xi Chanjuan,Yan Xiaoyu,You Wei,et al.Coupling reactions of zirconate complexes induced by carbonyl compounds[J].Angew Chem Int Ed,2009,48(43):8120-8123.
[15]Zhang Wen,Werness J B,Tang Weiping.Intramolecular hydroamination of conjugated enynes[J].Tetrahedron,2009,65(16):3090-3095.
[16]Zhou Yebing,Chen Jingjin,Zhao Chunbin,et al.Stereoselective synthesis ofβ-hydroxyallenes with multiple contiguous stereogenic centers via aldehyde addition toα-alkenyl-substituted zirconacyclopentenes[J].J Org Chem,2009,74(15):5326-5330.
[17]Ji Kegong,Shu Xingzhong,Zhao Shuchun,et al.Novel carbon-carbon bond formation from propargylic alcohols and olefin toward five-membered heterocyclic rings catalyzed by AgSbF6[J].Org Lett,2009,11(15):3206-3209.
[18]Azizoglu Akin,Balci Metin,Mieusset Jean-Luc,et al.Substituent effects on the ring-opening mechanism of lithium bromocyclopropylidenoids to allenes[J].J Org Chem,2008,73(21):8182-8188.
[19]吴阳锋,郑卫新,郑芬芬,等.1,2-丙二烯型化合物的合成[J].杭州师范大学学报:自然科学版,2010,9(2):113-117.
[20]Kippo T,Fukuyama T,Ryu I.Regioselective radical bromoallylation of allenes leading to 2-bromo-substituted 1,5-dienes[J].Org Lett,2011,13(15):3864-3867.
[21]Lin Li,Zhao Qiangyang,Li A-ni,et al.Enantioselective synthesis of Anomala osakana pheromone and Janus integer pheromone:a flexible approach to chiralγ-butyrolactones[J].Org Biomol Chem,2009,7(18):3663-3665.
[22]Sheldrake H M,Wallace T W.Reduction of propargylic sulfones to(Z)-allylic sulfones using zinc and ammonium chloride[J].Tetra-hedron Lett,2007,48(25):4407-4411.
[23]Wu Zhimeng,Huang Xian.Intermolecular Mn(OAc)3-mediated oxidative free-radical reaction of dimethyl malonate or ethyl cyanoacetate with allenes:An efficient synthesis of furan-2(5H)-ones or dimethyl 2-(2-oxoethylidene)malonates[J].Synthesis,2007(1):45-50.
[24]Al-Masum M,Yamamoto Y.Palladium-catalyzed hydrocarboxylation of allenes[J].J Am Chem Soc,1998,120(15):3809-3810.
[25]Yamazaki S,Yamamoto Y,Fukushima Y,et al.Lewis acid promoted reactions of ethenetricarboxylates with allenes:synthesis of indenes andγ-lactones via conjugate addition/cyclization reaction[J].J Org Chem,2010,75(15):5216-5222.
Synthesis of 1,1-Disubstituted-1,2-Propadiene
HU Lin-feng1,WANG Ping1,Zhang Yan-jing1,HONG Ya1,CHEN Cong1,MAO Guo-liang2,ZHENG Wei-xin1
(1.College of Materials and Chemical Engineering,Hangzhou Normal University,Hangzhou 310036,China;2.College of Chemistry and Chemical Engineering,Northeast Petroleum University,Daqing 163318,China)
The 1,1-disubstituted-1,2-propadiene was synthesized via SN2’reaction of Grignard reagent with sulfonic propargyl ester using alkyne as the raw material.The method has the advantages of readily available raw materials,mild conditions,few side reaction,high yields,et al.All products were verified by1H NMR,13C NMR,IR and MS.
1,1-disubstituted-1,2-propadiene;sulfonic propargyl ester;Grignard reagent;SN2’reaction;sy nthesis
O627
A
1674-232X(2012)04-0346-06
2012-02-03
国家自然科学基金项目(20972037);浙江省重点科技创新团队建设项目(2010R50017);杭州师范大学优秀中青年教师支持计划项目(HNUEYT 2011-01-013);黑龙江省留学归国人员科学基金项目(41417837-8-08016);黑龙江省教育厅海外学人科研资助项目(1154H14).
郑卫新(1975—),女,副教授,博士,主要从事有机合成研究.E-mail:wxzheng@hznu.edu.cn
11.3969/j.issn.1674-232X.2012.04.012
猜你喜欢
农药科学与管理(2019年8期)2019-11-23
石油炼制与化工(2017年1期)2017-04-06
赣南师范大学学报(2016年3期)2016-07-18
石油化工(2015年9期)2015-08-15
中国生化药物杂志(2015年4期)2015-07-07
湖南师范大学自然科学学报(2015年2期)2015-02-27
化学工业与工程(2015年1期)2015-02-10
中国药业(2014年21期)2014-05-26
石油炼制与化工(2014年4期)2014-04-06
郑州大学学报(理学版)(2014年4期)2014-03-01
