
荨麻疹性血管炎
2012-12-04 08:31吴宁俊田雅兰王官清
中国中西医结合皮肤性病学杂志 2012年6期
吴宁俊,田雅兰,王官清
(福建省厦门大学附属中山医院,厦门361004)
荨麻疹性血管炎(urticarial vasculitis,UV)又称血管炎性荨麻疹,是一种皮损似荨麻疹但组织病理上呈白细胞碎裂性血管炎改变的临床病理症候群,以单个皮损持续超过24 h,消退后留下出血点或色素沉着斑为特征。严重者伴关节痛、低补体血症,甚至多系统受累[1]。
1 分类
按血清补体水平分为正常补体血症性UV(normocomplementemic UV,NUV)和低补体血症性UV(hypocomplementemic UV,HUV)。但UV的皮损、实验室检查、组织病理等呈一病谱性改变,轻者有慢性特发性荨麻疹皮损但血清补体正常,称NUV;伴低补体血症者称HUV;HUV伴系统性红斑狼疮(SLE)、干燥综合征、冷球蛋白血症等基础疾病且抗C1q抗体阳性者称HUV综合征(HUVS),为最严重类型[2]。因此,NUV、HUV和HUVS代表UV进展过程,也有认为三者系独立疾病。UV还有一些特殊类型如关节炎-荨麻疹-血管性水肿(AHA)综合征、Schnitzler综合征、Cogan综合征和 Muckle-Wells 综合征[3-6]。
2 病因及发病机制
多数病因不明,称特发性UV。文献报道的诱因有自身免疫病、感染、药物、肿瘤等。自身免疫病以SLE和干燥综合征最常见,2%NUV和50%HUV被最终证实为SLE,32%UV伴干燥综合征,系统性硬化症[7]、混合性结缔组织病[8]、皮肌炎等也有报道。感染诱因包括甲肝、乙肝和丙肝病毒、EB病毒、肺炎支原体、螺旋体、弓形虫等。药物诱因包括培美曲塞、帕罗西汀、氟西汀、依那西普、英夫利昔单抗(infliximab)[9]、H1N1疫苗[10]等。相关肿瘤包括何杰金病、非何杰金淋巴瘤、单克隆丙种球蛋白病[4]等。少见诱因包括遗传性C3、C4缺乏症、C3肾炎因子缺乏症、炎性肠病[11]、运动[12]、寒冷、日光等。UV诱因存在地区差异,如泰国UV诱因依次为感染(12.5%)、药物(7.8%)、恶性肿瘤(7.8%)和 SLE(1.6%)[13]。
UV被认为是免疫复合物沉积血管壁所致,即Ⅲ型超敏反应。支持证据包括30%~75%患者循环免疫复合物(CIC)阳性、低补体血症、血浆置换可使皮损消退、皮损血管壁免疫球蛋白和补体沉积等。抗体可通过自身抗原和外源抗原诱导产生,其中抗C1q抗体、抗双链DNA(dsDNA)抗体、抗内皮细胞抗体(aECA)参与UV发病。抗C1q抗体是针对C1q胶原样区(collagen-like region)的低分子量(7S)IgG型自身抗体,与C1q结合形成C1q-抗C1q复合物后以Fc段依赖模式激活补体经典途径,使血清总补体(CH50)和C1q明显降低[14]。几乎所有HUVS均检出血清抗C1q抗体,多数SLE也检出抗C1q抗体,但尚无其参与UV的直接证据,可能仅为HUVS和SLE的相对特异抗体。已知抗dsDNA抗体参与肾小球肾炎发病,抗C1q抗体通过与dsDNA-抗dsDNA复合物中C1q结合使复合物可溶性降低而沉积损伤肾小球。aECA与内皮细胞结合也激活补体产生C3a、C5a和C5b-9损伤血管内皮,导致血管通透性增加、红细胞外渗、血管周围炎细胞浸润、甚至内皮细胞坏死和血栓形成,出现临床所见风团、血管性水肿、紫癜等皮损。而且,C1q可与血管内皮结合而成为自身抗原激发产生自身抗体[15]。在一项运动诱发UV实验中,分别在第3 h、10 h和24 h做连续皮肤病理切片,结果显示UV发病早期(3 h时)即有免疫复合物沉积、E-选择素和血管细胞黏附分子强表达、以及肥大细胞活化并释放肿瘤坏死因子-α,随后嗜酸粒细胞涌入释放颗粒蛋白,10 h时嗜中性粒细胞涌入并成为24 h时炎性浸润的主要细胞,提示嗜酸粒细胞颗粒蛋白在荨麻疹向血管炎进展过程中起关键作用[12]。
3 临床表现
3.1 发病率 慢性特发性荨麻疹中2%~20%最终被证实为UV。NUV发病高峰年龄约40岁,女性占60%~80%。HUV少见,经病理证实UV中仅18%为HUV;HUVS发病高峰年龄约 50 岁,男∶女=1∶2;儿童 UV 少见,约 2/3 为女性[16-18]。
3.2 皮损 可发生于全身任何部位,尤其受压部位。最常见皮损为风团,40%~60%患者仅有风团[16-18],经数天后可扩大形成斑块。42%~50%患者表现为血管性水肿,常为HUVS首发症状[18]。单个皮损持续超过24 h,常达3~4 d。多数皮损自觉瘙痒,但1/3患者感烧灼、疼痛和触痛[16]。皮损消退后留下紫癜或色素沉着斑,借助放大镜或皮肤镜观察更清楚。在皮肤镜下,红斑中央可见先前并不明显的紫癜、暗红色或褐色淤斑[19]。少见皮损包括环状红斑、多形红斑、匐行性回状红斑、网状青斑、雷诺现象。严重者出现紫癜、出血性水疱,甚至溃疡、坏死等[16-18]。
3.3 系统受累 HUV和HUVS易发生系统损害,关节、肺、胃肠道和肾脏常受累。
3.3.1 关节受累 是最常见的系统损害,与疾病活动性平行。多表现为关节痛或关节炎,约50%伴关节痛,关节炎为短暂性和游走性,手、肘、足、踝、膝等周边关节易受累[16]。HUV伴Jaccoud关节病预示心脏瓣膜病发生率增加[20]。
3.3.2 肺部受累 约20%UV伴肺部受累,吸烟者症状更重,包括胸膜炎、肺气肿、慢性阻塞性肺病(COPD)和哮喘,少见胸腔积液[16-17]。5%NUV和17%~20%HUV伴COPD和哮喘,50%HUVS伴中重度肺气肿性COPD,是HUVS常见死因之一。肺气肿可能因浸润的中性粒细胞和巨噬细胞释放弹性蛋白酶引起肺泡弹力纤维水肿、肺组织破坏所致[18]。
3.3.3 胃肠道受累 约17%~30%UV伴胃肠道症状,包括腹痛、恶心、呕吐、腹泻以及浆膜炎所致的腹水和肝脾肿大,少见胃肠道出血或缺血。
3.3.4 肾脏受累 约10%UV和20%~30%HUV伴肾脏受累[16],但症状轻微,表现为蛋白尿、镜下血尿,肾活检显示增生性肾小球肾炎、肾小管间质性肾炎、局灶坏死性肾炎等,但多为非进行性[18]。儿童HUV肾脏受累症状更重。
3.3.5 其他系统受累 约10%UV和30%HUVS伴眼部受累如结膜炎、巩膜炎、虹膜炎、葡萄膜炎、视神经盘或视网膜血管炎,甚至眼球萎缩。心血管受累包括心包炎、心包积液、心瓣膜病、雷诺现象等。神经系统受累包括假性脑瘤、癫痫、非化脓性脑膜炎、急性横断性脊髓炎、低位颅神经麻痹、周围神经病变等,假性脑瘤可能是静脉窦炎使脑脊液产生与吸收失平衡所致。肌肉活检示慢性肌炎,但无肌病症状。约10%患者伴发热、疲乏等全身症状[16]。
3.4 实验室检查 常见血沉(ESR)加快、补体C3和 C4降低、自身抗体阳性、血尿等实验室检查异常。
3.4.1 ESR加快 见于29%NUV和50%HUV,为非特异性指标,与病情严重度或有无系统受累无关[17]。
3.4.2 低补体血症 是公认的系统受累和并发症标记。18%~32%UV血清CH50降低,HUVS血清C3、C4和C1q明显降低,病情缓解后C3、C4水平恢复正常时C1q仍持续降低[1,18]。
3.4.3 自身抗体阳性 27%NUV和71%HUV呈抗核抗体(ANA)低滴度阳性,24%UV呈抗dsDNA阳性,29.2%HUV呈浸出性核抗原多肽(ENA,包括ssA和Sm)抗体阳性。NUV仅与干燥综合征或SLE重叠时才出现抗ssA和ssB阳性。HUVS呈ANA阴性或中等滴度阳性,但抗DNA抗体和抗Sm抗体阳性率超过95%。14%NUV、82%SLE合并UV和70%HUVS呈aECA阳性,几乎所有HUVS呈C1q抗体阳性。偶见抗磷脂抗体、抗甲状腺抗体阳性。
3.4.4 其他 外周血白细胞升高、轻度贫血。蛋白尿、脓尿和血尿,出现蛋白尿者均为HUV。12%UV类风湿因子(RF)阳性。部分患者CIC阳性,血清IgA、IgG尤其是IgG4升高[21]。C1脂酶抑制剂含量正常但无功能[22]。
3.5 组织病理和免疫病理 UV主要累及小静脉,应更准确称为荨麻疹性小静脉炎。组织病理上呈白细胞碎裂性血管炎改变,包括血管内皮细胞肿胀,血管壁纤维素样变,小静脉内及周围嗜中性粒细胞、单核细胞和嗜酸粒细胞浸润,血管周围白细胞碎裂(核尘)和红细胞外渗,严重者血栓形成甚至血管破坏。血管壁纤维素样变、血管周围核尘和红细胞外渗是血管损伤的直接证据,纤维素样变或核尘是诊断UV的最基本组织病理标准。因部分UV合并慢性荨麻疹,两者组织病理改变存在连续性,从仅有血管周围炎细胞浸润到明显的血管炎改变反映UV皮损的进展过程[23]。因此有认为毛细血管和毛细血管后静脉内或静脉周围的混合性炎细胞浸润即足以诊断UV,即便无纤维素样变或核尘[1,16-17]。炎性浸润细胞中NUV以嗜酸粒细胞为主,HUV以嗜中性粒细胞为主。直接免疫荧光(DIF)检查,超过70%UV尤其活动期皮损基底膜带(BMZ)和血管壁免疫球蛋白(IgM、IgG和 IgA)、补体 C3和(或)纤维素沉积。HUV患者IgG和(或)补体C3沉积预示其发展为SLE或肾脏疾病可能[17]。
Peroni等[24]总结了近20年文献报道的NUV和HUV临床特点比较,见表1。
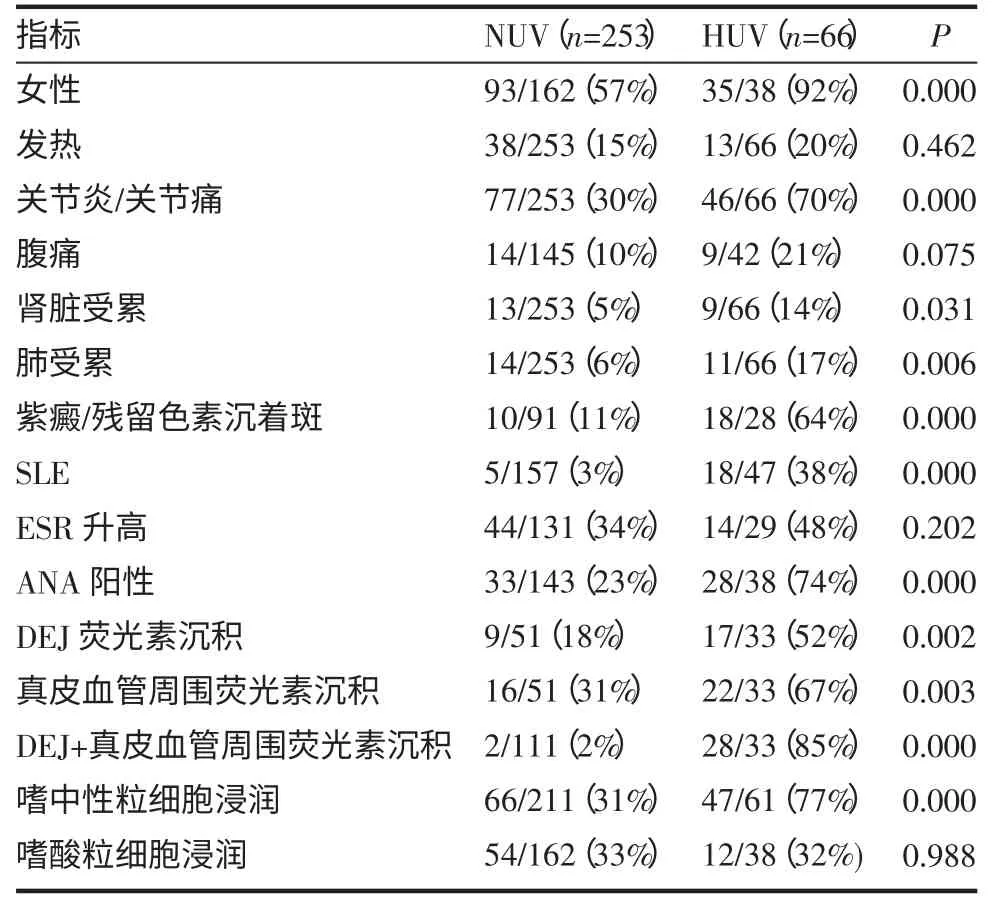
表1 NUV和HUV主要临床特点比较例(%)
4 诊断和鉴别诊断
对荨麻疹而言首先应确定为急性或慢性,当反复或持续发作达6周或以上即可诊断慢性荨麻疹。通过自体血清实验、食物假性过敏原口服激发实验、放射性变应原吸附试验、感染灶检查、肿瘤筛查等确定其病因为自体反应性、食物不耐受性、药物性、感染性或副肿瘤性。如荨麻疹持续超过24 h、伴烧灼或疼痛感、消退后留下出血点或色素沉着斑,应高度怀疑UV并取12 h以内早期皮损做组织病理和免疫荧光检查[25]。诊断确立后需结合详尽病史、体格检查及必要的实验室检查如ANA、抗ENA抗体、抗C1q抗体、冷球蛋白、补体、全血细胞计数、Schirmer试验、肾脏和肺功能测定等确定UV是独立疾病或某综合征[1]。按血清补体水平进行UV分类需在数月内检测血清CH50、C3、C4和C1q水平2~3次,在UV平稳期和活动期内补体均正常时才能诊断NUV。若伴血清补体降低,尤其UV复发时,可诊断HUV。少数诊断为原发性或特发性HUV者如最终证实伴SLE、干燥综合征、冷球蛋白血症等基础疾病,抗C1q抗体阳性,即可诊断HUVS[1]。
4.1 UV的诊断和鉴别诊断 荨麻疹反复发生,单个风团持续超过24 h,伴灼热或疼痛,消退后留下瘀斑或色素沉着斑,有白细胞碎裂血管炎组织病理依据即可诊断UV[26]。与慢性荨麻疹鉴别见表2[2],临床上可采用玻片压诊试验和皮肤镜观察鉴别。玻片压诊试验是将玻片压于疑似UV皮损上,如红斑消退但中央紫癜不退应高度怀疑UV。皮肤镜下UV皮损中央的红色出血点透露其血管炎本质[19,27]。
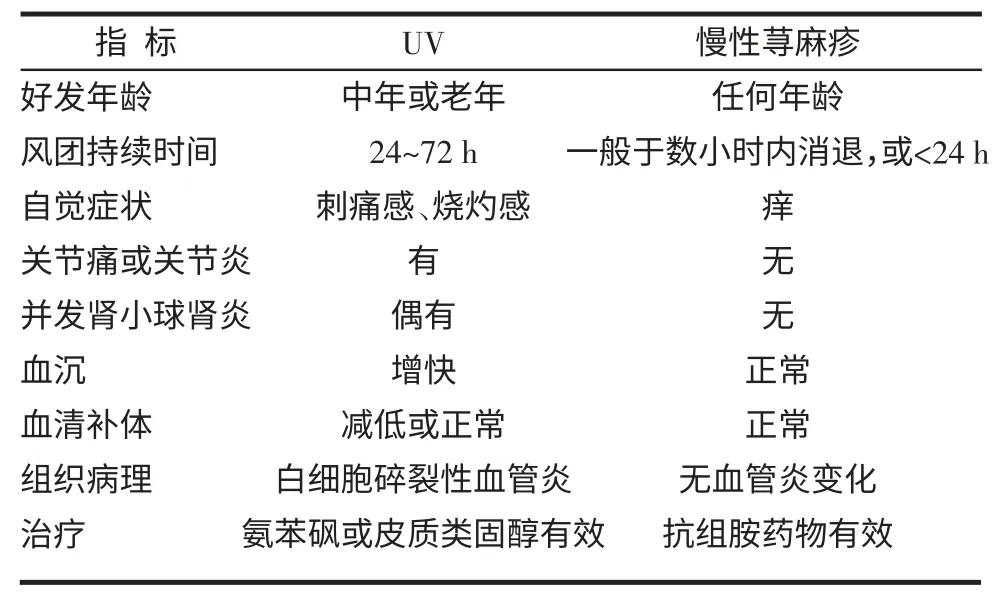
表2 UV与慢性荨麻疹鉴别要点
4.2 HUVS的诊断和鉴别诊断 HUVS诊断主要指标:(1)慢性荨麻疹皮损;(2)低补体血症。次要指标:(1)白细胞碎裂性血管炎;(2)关节痛或关节炎;(3)葡萄膜炎、巩膜炎(或结膜炎);(4)肾小球肾炎;(5)腹痛;(6)C1q 抗体阳性。除典型慢性荨麻疹皮损外,关键是低补体血症和白细胞碎裂性血管炎的组织病理证据。血清补体水平是判断病情严重度重要指标,C3、C4和C1q均显著降低,抗C1q抗体为非特异指标[2]。HUVS需与SLE、混合性冷球蛋白血症鉴别,50%HUVS呈ANA阳性并符合SLE诊断标准,三者活动期均有低补体血症和多系统受累,但高滴度抗dsDNA抗体或抗Sm抗体可排除HUVS,混合性冷球蛋白血症的冷沉淀中单克隆免疫球蛋白和高浓度冷球蛋白呈阳性[2]。
5 治疗
目前缺乏严格对照的UV治疗研究,也无一种治疗措施普遍有效[1-2],治疗措施取决于病情严重度。
5.1 抗组胺药物 治疗轻症UV的主要药物,可减轻瘙痒症状但不能改变病程。H1受体拮抗剂很少单独奏效,可联用H2受体拮抗剂、酮替芬或多虑平[18]。
5.2 氨苯砜 单用氨苯砜可使皮损消退,但需注意其不良反应。严重病例可联用皮质类固醇激素或免疫抑制剂。
5.3 皮质类固醇激素 治疗系统性损害的主要药物,HUV伴系统性损害者及早应用可预防肾损害[1,18]。初始剂量一般相当于强的松1 mg/(kg·d),分次口服或缓慢静脉滴注,待体温恢复正常、皮损消退后逐渐减量维持。但停用后易复发,需长期维持。因UV病程较长,可考虑地塞米松5 mg/d静脉滴注或2.5 mg口服,2 次/d,每周连用 3 d。
5.4 免疫抑制剂 严重病例可联用免疫抑制剂如硫唑嘌呤、环磷酰胺、环孢素、吗替麦考酚酯(mycophenolate mofetil)[27]等以减少激素的用量和不良反应。有报告地塞米松和环磷酰胺冲击治疗HUV取得较好疗效,方法是环磷酰胺500 mg和地塞米松100 mg静脉滴注,1次/d,3 d为1个疗程,2~4周重复1个疗程,间歇期口服环磷酰胺50 mg/d,16疗程后停药。环孢素A可明显减轻HUVS患者慢性阻塞性肺病(COPD)气道阻塞症状,但甲氨喋呤效果不佳并可能加重病情[28]。
5.5 血浆置换 常用于高度活动患者,可降低CIC,但作用短暂,停用数天症状常复发,联用免疫抑制剂以阻止抗C1q抗体产生则治疗效果更佳,静脉注射丙种球蛋白也有效。
5.6 其他 非甾体类抗炎药(NSAID)如消炎痛、芬必得、诺松等可试用。秋水仙碱和抗疟药可减少皮质类固醇激素用量。UV伴丙肝病毒感染和冷球蛋白血症者联用α-2b干扰素和利巴韦林治疗可使50%患者病毒感染受抑制、皮损消退,但停药易反跳[18]。IL-1受体拮抗剂阿那白滞素(anakira)治疗NUV、Schnitzler综合征和Muckle-Wells综合征有效[6,29],抗B细胞抗体利妥昔单抗(rituximab)疗效尚待评估[30]。有学者建议治疗UV时可依次选用抗组胺药、NSAID、秋水仙碱、氨苯砜或羟氯喹、皮质类固醇和免疫抑制剂。
6 病程及预后
UV病程最长达23年,平均3~4年,皮损可呈反复加重、消退和复发。特发性NUV预后良好,HUV和HUVS预后较差,主要死因为COPD和急性喉头水肿,少见死于原发病[18]。
[1]Puig L.Urticaria vasculitis[A].in Zuberbier T,Grattan CEH.Mau rer M.Urticaria and angioedema[M].Springer-Verlag Berlin Heidelberg,2010:109-116.
[2]Grotz W,Babe HA,Becker JU,et al.Hypocomplementemic uriti caria vasculitis syndrome,an interdisciplinary challenge[J].Dtsch Arztebl Int,2009,106:756-763.
[3]Mc Neil DJ,Kinsella TD,Craford AM,et al.The AHA syndrome:arthritis,hives and angioedema[J].Rheumatol Int,1987,7:277-279.
[4]Carlesimo M,Abruzzese C,Narcisi A,et al.Chronic vasculitis urticaria associated to a monoclonal gammopathy of IgM and IgA type,a Schnitzler syndrome[J].Eur J Dermatol,2010,20:838-839.
[5]St ClairEW,McCallumRM.Cogan’ssyndrome[J].CurrOpinRheumatol,1999,11:47-52.
[6]Hawkins PN,Lachmann HJ,Agama E,et al.Spectrum of clinical features in Muckle-Wells syndrome and response to anakinra[J].Arthritis Rheum,2004,50:607-612.
[7]Kato Y,Aoki M,Kawana S.Urticarial vasculitis appearing in the progression of systemic sclerosis[J].J Dermatol,2006,33:792-797.
[8]Calistru AM,Lisboa C,Cruz MJ,et al.Hypocomplementemic ur ticarial vasculitis in mixed connective tissue disease[J].Dermatol Online J,2010,16:8.
[9]Goulo J,Cunha H,Anes I,et al.Urticarial vasculitis due do infliximab[J].J Eur Acad Dermatol Venereol,2008,22:882-883.
[10]Hughes R,Lacour JP,Baldin B,et al.Urticarial vasculitis secondary to H1N1 vaccination[J].Acta Derm Venereol,2010,90:651-652.
[11]Swaminath A,Magro CM,Dwyer E.Refractory urticarial vasculitis as a complication of ulcerative colitis successfully treated with rituximab[J].J Clin Rheumatol,2011,17:281-283.
[12]Kano Y,Orihara M,Shiohara T.Cellular and molecular dynamics in exercise-induced urticarial vasculitis lesions[J].Arch Dermatol,1998,134:62-67.
[13]Kulthawee K,Cheepsomsong M,Jiamton S.Urticairial vasculitis,e-tiologies and clinical course[J].Asian Pacific J Allergy Immunol,2009,27:95-102.
[14]Wisnieski JJ,Jones SM.IgG autoantibody to collagen-like region of C1q in hypocomplementemic urticarial vasculitis syndrome,systemic lupus erythematosus,and 6 other musculoskeletal or rheumatic diseases[J].J Rheumatol,1992,19:884-888.
[15]O’Flynna J,Fliermana R,van der Pola P,et al.Nucleosomes and C1q bound to glomerular endothelial cells serve as targets for autoantibodies and determine complement activation[J].Mol Immunol,2011,49:75-83.
[16]Mehregan DR,Hall MJ,Gibson LE.Urticarial vasculitis:a histopathologic and clinical review of 72 cases[J].J Am Acad Dermatol,1992,26:441-448.
[17]Davis MD,Daoud MS,Kirby B,et al.Clinicopathologic correlation of hypocomplementemic and normocomplementemic urticarial vasculitis[J].J Am Acad Dermatol,1998,38:899-905.
[18]Davis MD,Brewer JD.Urticarial vasculitis and hypocomple -mentemic urticarial vasculitis syndrome[J].Immunol Allergy Clin North Am,2004,24:183-213.
[19]Vázquez-López F,Fueyo A,Sánchez-Martín J,et al.Dermoscopy for the screening of common urticaria and urticaria vasculitis[J].Arch Dermatol,2008,144:568.
[20]Amano H,Furuhata N,Tamura N,et al.Hypocomplementemic urticarial vasculitis with Jaccoud's arthropathy and valvular heart disease,case report and review of the literature[J].Lupus,2008,17:837-841.
[21]Wakamatsu R,Watanabe H,Suzuki K,et al.Hypocomplementemic urticarial vasculitis syndrome is associated with high levels of serum IgG4:a clinical manifestation that mimics IgG4-related disease[J].Int Med,2011,50:1109-1112.
[22]Moulin C,Debarbieux S,Ducastelle-Lepretre S,et al.Urticarial vasculitis and asymptomatic acquired C1 esterase inhibitor deficiency revealing an angioimmunoblastic T cell lymphoma[J].Eur J Dermatol,2010,20:515-516.
[23]Jones RR,Bhogal B,Dash A,et al.Urticaria and vasculitis:a continuum of histological and immunopathological changes[J].Br J Dermatol,1983,108:695-703.
[24]Peroni A,Colato C,Zanoni G,et al.Urticarial lesions:if not urticaria,what else?The differential diagnosis of urticaria[J].J Am A-cad Dermatol,2010,62:557-570.
[25]李晓霞,王合,王德辉,等.荨麻疹性皮肤血管炎15例临床病理分析 [J].中国皮肤性病学杂志,2008,22(12):726-728.
[26]Tosoni C,Lodi-Rizzini F,Cinquini M,et al.A reassessment of diagnostic criteria and treatment of idiopathic urticarial vasculitis:a retrospective study of 47 patients[J].Clin Exp Dermatol,2009,34:166-170.
[27]Lee JS,Loh TH,Seow SC,et al.Prolonged urticaria with purpura:the spectrum of clinical and histopathologic features in a prospective series of 22 patients exhibiting the clinical features of urticarial vasculitis[J].J Am Acad Dermatol,2007,56:994-1 005.
[28]Worm M,Sterry W,Kolde G.Mycophenolate mofetil is effective for maintenance therapy of hypocomplementemic urticarial vasculitis[J].Br J Dermatol,2000,143:1324.
[29]Botsios C,Sfriso P,Punzi L,et al.Non-complementaemic urticarial vasculitis:successful treatment with the IL-1 receptor antagonist,anakinra[J].Scand J Rheumatol,2007,36:236-237.
[30]Mukhtyar C,Misbah S,Wilkinson J,et al.Refractory urticarial vas culitis responsive to anti-B-cell therapy[J].Br J Dermatol,2009,160:470-472.
猜你喜欢
开卷有益·求医问药(2022年8期)2022-09-29
现代医院(2022年6期)2022-08-26
家庭科学·新健康(2021年9期)2021-09-17
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年2期)2021-03-29
河南医学研究(2021年4期)2021-03-10
保健与生活(2020年14期)2020-07-27
家庭科学·新健康(2019年5期)2019-06-06
中国医药导报(2018年12期)2018-06-20
中国医药指南(2017年3期)2017-11-13
