
硫脲催化硝基烯与硫叶立德的不对称Michael加成反应
2012-11-30 10:33孙香婷张冬菊冯大诚刘成卜
物理化学学报 2012年3期
孙香婷 张冬菊 冯大诚 刘成卜
(山东大学化学与化工学院,理论化学研究所,济南250100)
硫脲催化硝基烯与硫叶立德的不对称Michael加成反应
孙香婷 张冬菊*冯大诚 刘成卜
(山东大学化学与化工学院,理论化学研究所,济南250100)
基于密度泛函理论研究了氯代苯基硫脲催化的硝基苯乙烯与硫叶立德的Michael加成反应,确定了控制反应立体选择性的C―C键形成步骤的过渡态结构,计算了过渡态的相对能量和反应势垒,弄清了硫脲催化的微观反应机理,探讨了硫脲催化性能的微观本质.结果表明,反应有利于反式Michael加成产物的形成,硫脲在反应中作为质子给体,首先与质子受体硝基苯乙烯形成双氢键配合物,通过授受体间的电荷转移活化硝基苯乙烯的β-C原子增强其亲电性,有利于硫叶立德的亲核进攻.
硫脲;硝基苯乙烯;硫叶立德;不对称Michael加成;密度泛函理论
1 引言
Michael加成反应是构建C―C骨架的有效方法之一,在有机合成领域有重要的应用价值.近年来,有机小分子催化不对称Michael加成反应日益引起广泛关注,已成为合成众多手性分子和药物中间体的有效手段.1-3脯氨酸及其衍生物、手性咪唑啉衍生物、金鸡纳碱及其衍生物、手性胍、手性(硫)脲、多肽和手性离子液体等有机催化剂已被成功用于不对称Michael加成反应,获得了高收率、高对映选择性的产物.4-8
不饱和羰基化合物与硝基烯化合物的Michael加成反应是制备硝基烷烃,并进而制备胺、羰基化合物、羧酸等化合物的重要方法,在不对称Michael加成反应中应用非常广泛.多年来,人们一直致力于优化这类反应的催化剂,改进反应条件,扩大底物的适应范围等,虽已取得了一些突破性进展,但在缩短反应时间、提高反应产率和增加产物光学活性等方面尚未取得令人满意的进步.
最近,Xiao等9报道了硫脲(thiourea)催化的硝基烯(nitroolefin)与硫叶立德(sulfur ylide)的Michael加成反应,如图1所示.实验发现,通过该反应获得的Michael加成产物的产率高达95%,其中反式和顺式产物的非对映选择性之比最高可达99:1.
为在分子水平上理解这类硫脲催化的Michael加成反应的微观机理,寻找催化剂活化反应底物、控制反应立体选择性的微观本质,本文以图2所示的反应为例,基于密度泛函理论计算,研究控制产物立体选择性的关键步骤,即C―C键形成步骤,探讨详细的微观机理,分析影响产物立体选择性的微观本质.
2 模型体系与计算方法
本文研究的模型体系如图2所示,使用一个典型的硫叶立德作为亲核试剂,硝基苯乙烯作为亲电底物,氯代苯基硫脲为催化剂.
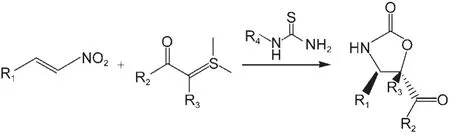
图1 硫脲催化硝基烯与硫叶立德的不对称Michael加成反应9Fig.1 Asymmetric Michael addition between nitroolefins and sulfur ylides catalyzed by thioureas9
本文的理论计算基于密度泛函理论,研究该反应关键步骤的微观机理,计算反应的热力学和动力学性质.由于实验上该反应是在二氯甲烷溶剂中进行的,溶剂化效应对反应机理影响不大,因此我们使用气相模型进行研究.计算中使用杂化的B3LYP泛函10,11和6-31G(d,p)标准基组对反应物、催化剂、过渡态、C―C键加成产物等进行了全构型优化和振动频率分析,对过渡态结构进行了内禀反应路径追踪,以确保获得势能剖面上真正的稳定结构、一级鞍点和最低反应途径.全部计算使用Gaussian 03程序,12在山东大学理论化学所高性能计算中心完成.
3 结果与讨论
3.1 氯代苯基硫脲、硝基苯乙烯和硫叶立德的结构
图3给出了氯代苯基硫脲、硝基苯乙烯和硫叶立德的优化构型和关键的键长键角数据.括号内是相应的晶体结构数据.计算的结构参数与实验数据符合较好,表明B3LYP泛函和中等尺寸的6-31G(d, p)基组可以较好地描述所研究的反应体系.中等水平的理论方法(B3LYP/6-31G(d,p))被广泛用于研究有机小分子催化的不对称C―C加成反应的立体选择性.13,14
氯代苯基硫脲是一个准平面型分子,分子内存在弱的氢键相互作用,硫脲的两个N―H键与苯环上的氯位于环的同一侧,与晶体结构测定结果一致.硝基苯乙烯基态呈反式构型,具有Cs对称性,是一个平面型分子,其中硝基氧与C=C双键上的H原子形成弱氢键.硫叶立德呈C1对称性,羰基氧与临近的三个氢原子形成3个弱的氢键.

图2 氯代苯基硫脲催化硝基苯乙烯与硫叶立德的不对称Michael加成反应Fig.2 Asymmetric Michael addition between trans-β-nitrostyrene and a typical sulfur ylide catalyzed by 1-(2-chlorophenyl)-2-thiourea
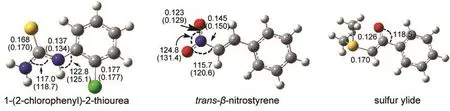
图3 B3LYP/6-31G(d,p)水平上优化的催化剂和反应物构型及其结构参数Fig.3 Geometries of reactants and catalyst with selected structural parameters optimized at the B3LYP/6-31G(d,p)level Distances are in nm,angles are in degree,and the values in parentheses are experimental findings.
3.2 氯代苯基硫脲-硝基苯乙烯二元配合物
催化剂与反应底物的结合是反应的初始步骤, Zheng等15-17曾报道过硫脲与硝基化合物的结合方式,发现硫脲化合物与硝基化合物之间可以形成强的氢键,我们的计算也进一步验证了前人的研究结果.图4给出了优化的二元复合物的结构,我们观察到硫脲中两个N―H键的氢原子距离(0.218 nm)与硝基苯乙烯中两个硝基氧原子距离(0.219 nm)相近,前者是质子给体,而后者容易接受质子,由此可以推测催化剂将优先与硝基苯乙烯形成双氢键配合物,以固定和活化反应底物.图4中氯代苯基硫脲与硝基苯乙烯形成的两个二元配合物是结构相似的配合物,硝基苯乙烯双键分别位于硫脲苯环的异侧和同侧,硫脲的两个N―H基团作为氢键给体、硝基苯乙烯的两个氧原子作为氢键受体形成双氢键配合物,计算的结合能分别是42.1和41.5 kJ·mol-1,N―H··O氢键距离在0.21-0.22 nm之间.较大的结合能和有效的氢键距离表明氢键配合物具有较高的稳定性.Mulliken电荷布居分析表明,两个配合物中β-双键碳原子所带的负电荷分别为-0.088e和-0.090e,与孤立的硝基苯乙烯中的-0.095e相比,亲电性明显增强,有利于硫叶立德碳负离子的亲核进攻,使得C―C键加成反应更容易进行.
另外,我们也尝试优化了氯代苯基硫脲与另一反应底物硫叶立德的配合物,但结果表明二者难于形成稳定的配合物.因此氯代苯基硫脲与硝基苯乙烯的结合应是反应的初始步骤.
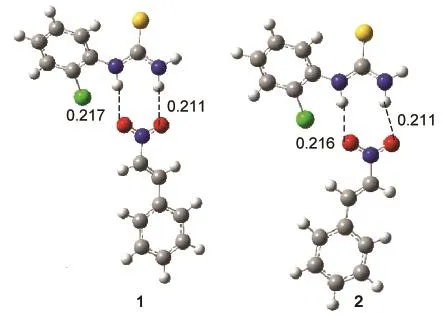
图4 硝基苯乙烯与氯代苯基硫脲形成的双氢键配合物Fig.4 Optimized double H-bonding complexes between trans-β-nitrostyrene and 1-(2-chlorophenyl)-2-thioureaDistances are in nm.
3.3 氯代苯基硫脲-硝基苯乙烯-硫叶立德三元配合物
稳定的氢键配合物的形成起到了固定和活化底物的双重作用,接下来的过程是硫叶立德对双氢键配合物的亲核进攻,形成氯代苯基硫脲-硝基苯乙烯-硫叶立德三元配合物.当硫叶立德进攻双氢键配合物的亲核中心时,考虑到它们前手性中心Re面和Si面(Re和Si分别表示前手性中心碳原子上三个基团的优先顺序是顺时针方向和逆时针方向)的不同组合,三元配合物存在四种立体异构体,分别表示为IMSi-Si,IMRe-Re,IMSi-Re和IMRe-Si,下标中前一个Si或Re表示双氢键配合物的Si或Re面,后一个Si或Re代表硫叶立德的Si或Re面.

图5 氯代苯基硫脲-硝基苯乙烯-硫叶立德三元配合物的构型、主要结构参数及相对能量Fig.5 Optimized ternary complexes among trans-β-nitrostyrene,1-(2-chlorophenyl)-2-thiourea,and the sulfur ylide with main structural parameters and relative energiesEreldenotes relative energies(in kJ·mol-1).Distances are in nm.
图5给出了优化的四个三元配合物的构型,其中Erel表示相对能量.这些三元配合物可以视为硫叶立德与二元配合物通过范德华作用形成的超分子结构,其中氯代苯基硫脲与硝基苯乙烯之间仍保持稳定的双氢键结构,而硫叶立德与二元配合物距离较远.四个异构体中,IMSi-Si是最稳定的,计算得到的稳定化能为32.2 kJ·mol-1.IMRe-Re的能量比IMSi-Si高4.8 kJ·mol-1,另外二个结构IMSi-Re和IMRe-Si的相对能量分别是15.2和31.6 kJ·mol-1.显然,IMSi-Si和IMRe-Re是其中的相对优势构型.
3.4 过渡态和产物
在三元配合物中,硫叶立德进一步接近硝基苯乙烯将导致C―C键的形成,该过程是控制Michael加成产物立体选择性的关键步骤,18-25也是本文所要重点关注和讨论的问题.与IMSi-Si,IMRe-Re,IMSi-Re和IMRe-Si四个三元配合物中间体相对应,我们确定了四个过渡态结构,标记为TSSi-Si,TSRe-Re,TSSi-Re和TSRe-Si,如图6所示,在这些过渡态结构中,正在形成的C―C键距离在0.21-0.22 nm之间,其虚频振动模式对应了C―C键的生成过程.
TSSi-Si是最稳定的过渡态,对应硫叶立德的Si面进攻双氢键配合物的Si面,该过程的能垒为69.6 kJ·mol-1,生成的C―C键加成产物中含两个手性碳原子(C2和C3),分别为S和R构型,标记为(2S,3R).由于该产物中苯基和苯羰基处于碳碳键的反式(anti)位置,得到的产物是反式产物,联合S和R构型的标记,在图7中标记为(2S,3R)-anti型产物.
TSRe-Re对应硫叶立德的Re面进攻双氢键配合物的Re面的过渡态,其能量仅比最稳定的TSSi-Si高3.3 kJ·mol-1,但该过程的能垒为68.1 kJ·mol-1,比形成过渡态TSSi-Si低1.5 kJ·mol-1,所以该路径与经过TSSi-Si的路径是竞争、共存的,其产物标记为(2R,3S)-anti,也是反式产物.
另外两个路径分别经过TSSi-Re和TSRe-Si,它们的相对能量分别比最稳定的过渡态高TSSi-Si高30.3和39.4 kJ·mol-1,对应的C―C键加成产物都为顺式产物,分别标示为(2S,3S)-syn和(2R,3R)-syn,其中syn表示加成产物的苯基和苯羰基在C―C键的同侧,两个碳碳加成过程的能垒分别是84.7和77.4 kJ·mol-1,明显高于形成反式产物的能垒,可以得出结论,两个形成顺式产物的路径是相对不利的.
图7列出了C―C键加成产物的四个立体异构体,(2S,3R)-anti,(2R,3S)-anti,(2S,3S)-syn和 (2R, 3R)-syn,分别对应着TSSi-Si,TSRe-Re,TSSi-Re和TSRe-Si四个过渡态对应的向前的产物,通过IRC计算和结构优化得到.
为了比较四条路径的相对能量,图8给出了计算得到的相对能量,由此可以清楚地看出TSSi-Si是能量最低的过渡态,形成的(2S,3R)-anti是最稳定的反式C―C加成产物,其基元步骤的能垒为69.6 kJ·mol-1.另一种反式C―C加成产物(2R,3S)-anti通过过渡态TSRe-Re形成,TSRe-Re相对能量比TSSi-Si高3.3 kJ·mol-1,但由于该过程的能垒(68.1 kJ·mol-1)比前者尚低1.5 kJ·mol-1,所以我们认为两个反应通道是竞争的,均贡献到反式产物的形成.顺式C―C加成产物的稳定性明显低于反式C―C加成产物,且需要跨越较高的能垒,因此是热力学和动力学上均不利的过程.
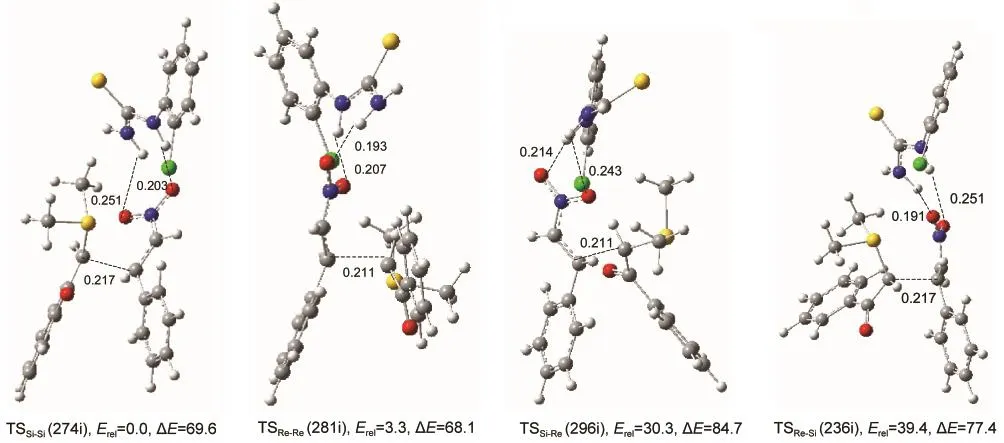
图6 不对称Michael加成C―C加成步骤的过渡态构型、主要结构参数及相对能量Fig.6 Optimized transition state geometries with the main structural parameters for the C―C bond-formation step of the asymmetric Michael additioni denotes the imaginary frequency,Ereldenotes relative energies,ΔE denotes the barriers to be overcome to reach the transition state. Energies are in kJ·mol-1,distances are in nm,frequencies are in cm-1.

图7 C―C加成步骤产物构型及其相对能量Fig.7 Optimized geometries and calculated relative energies of the C―C bond addition products Energies are in kJ·mol-1.

图8 计算得到的四条反应路径中C―C加成步骤的相对能量Fig.8 Energy diagrams of the C―C bonding-formation steps along the four pathways1 and 2 denote the two complexes shown in Fig.4.
Xiao等9在实验上得到该反应的反式与顺式产物之比是95:5,我们的计算结果可以合理解释产物的高非对映选择性.比较四个路径的过渡态构型,我们发现在生成顺式产物的两个过渡态中,硫叶立德的几何构型发生了较大扭曲,空间位阻较大,致使反应能垒升高,不利于反应的进行,因此顺式产物在最终产物所占比例较小.
C―C加成产物再经过一系列质子转移过程即可转变为最终的Michael加成产物,9这些质子转移过程对整个反应的立体选择性没有影响,且机理较为清晰,因此本文对后续的质子转移过程未做进一步讨论.
4 结论
基于密度泛函理论计算研究了氯代苯基硫脲催化的硝基苯乙烯与硫叶立德的Michael加成反应.结果表明,反应的初始步骤是氯代苯基硫脲通过两个氨基与硝基苯乙烯形成双氢键配合物,起到底物和活化底物的作用,氢键授受体之间的电荷转移增强硝基苯乙烯β-双键碳原子的亲电能力,有利于硫叶立德的亲核进攻;在C―C键形成的关键步骤中,双氢键复合物的Si面对硫叶立德Si面的进攻是最有利反应通道,反应能垒是69.6 kJ·mol-1,形成反式加成产物,而顺式加成产物的形成需要克服的最低能垒是77.4 kJ·mol-1,在能量上是不利的.本文的计算结果一方面合理解释了实验观测的高非对映选择性(反式产物/顺式产物为95/5),另一方面也阐明了硫脲催化剂的催化机制,合理地解释了实验现象,所得结果不仅可以加深人们对硫脲催化剂催化机制的认识,而且对硫脲为基础的新型有机催化剂的设计具有一定指导意义.
(1)Brown,S.P.;Goodwin,N.C.;MacMillan,D.W.C.J.Am. Chem.Soc.2003,125,1192.
(2) Enders,D.;Seki,A.Synlett 2002,26.
(3) Andrey,O.;Alexakis,A.;Bernardinelli,G.Org Lett.2003,5, 2559.
(4) Ishii,T.;Fujioka,S.;Sekiguchi,Y.;Kotsuki,H.J.Am.Chem. Soc.2004,126,9558.
(5) List,B.;Prjarliev,P.;Martin,H.J.Org.Lett.2001,3,2423.
(6) Okino,T.;Hoashi,Y.;Furukawa,T.;Takemoto,Y.J.Am.Chem. Soc.2005,127,119.
(7) Alexakis,A.;Andrey,O.Org.Lett.2002,4,3611.
(8) Betancort,J.M.;Barbas,C.F.Org.Lett.2001,3,3737.
(9) Lu,L.Q.;Cao,Y.J.;Liu,X.P.;An,J.;Yao,C.J.;Ming,Z.H.; Xiao,W.J.J.Am.Chem.Soc.2008,130,6946.
(10) Hay,P.J.;Wadt,W.R.J.Chem.Phys.1985,82,299.
(11) Hay,P.J.;Wadt,W.R.J.Chem.Phys.1985,82,270.
(12) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, Revision D.01;Gaussian Inc.:Wallingford,CT,2004.
(13) Fu,A.P.;Li,H.L.;Yuan,S.P.;Si,H.Z.;Duan,Y.B.J.Org. Chem.2008,73,5264.
(14) Fu,A.P.;Li,H.L.;Tian,F.H.;Yuan,S.P.;Si,H.Z.;Duan,Y. B.Tetrahedron:Asymmetry 2008,19,1288.
(15) Zheng,W.R.;Xu,L.J.;Huang,T.;Yang,Q.;Chen,Z.C.Res. Chem.Intermed.2011,37,31.
(16) Zheng,W.R.;Fu,Y.;Shen,K.;Liu,L.;Guo,Q.X.J.Mol. Struct:Theochem 2007,822,103.
(17) Zheng,W.R.;Fu,Y.;Liu,L.;Guo,Q.X.Acta Physico-Chimica Sinica 2007,23,1018.[郑文锐,傅 尧,刘 磊,郭庆祥.物理化学学报,2007,23,1018.]
(18) Hoashi,Y.;Okino,T.;Takemoto,Y.Angew.Chem.Int.Edit. 2005,44,4032.
(19) Xu,X.;Yabuta,T.;Yuan,P.;Takemoto,Y.Synlett 2006,136.
(20) Xu,X.;Furukawa,T.;Okino,T.;Miyabe,H.;Takemoto,Y. Chem.Eur.J.2006,12,466.
(21) Li,Q.;Huang,F.Q.Acta Physico-Chimica Sinica 2005,21,52. [李 权,黄方千.物理化学学报,2005,21,52.]
(22) Tan,B.;Zeng,X.F.;Lu,Y.P.;Chua,P.J.;Zhong,G.F.Org. Lett.2009,11,1927.
(23) Lu,N.;Meng,L.;Chen,D.Z.;Zhang,G.Q.J.Phys.Chem.A 2012,116,670.
(24)Yang,H.;Wong,M.W.J.Org.Chem.2011,76,7399.
(25) Marju,L.;Kerti,A.;Merle,U.;Toomas,T.;Tonis,K.;Margus, L.J.Org.Chem.2009,74,3722.
December 5,2011;Revised:January 4,2012;Published on Web:January 11,2012.
Asymmetric Michael Addition between Nitroolefins and Sulfur Ylides Catalyzed by a Thiourea Organocatalyst
SUN Xiang-Ting ZHANG Dong-Ju*FENG Da-Cheng LIU Cheng-Bu
(School of Chemistry and Chemical Engineering,Institute of Theoretical Chemistry,Shandong University, Jinan 250100,P.R.China)
Using density functional theory calculations,we have studied the 1-(2-chlorophenyl)-2-thiourea catalyzed reaction of nitrostyrene with a typical sulfur ylide to understand the Michael addition mechanism. Transition state structures for the C―C bond-forming step controlling the stereoselectivity of the reaction have been identified and their relative stabilities evaluated.The role of the catalyst in the reaction has also been determined.The calculated results show that the formation of the anti-product is energetically more favorable than that of the syn-product.Furthermore,the catalyst(proton donor)promotes the reaction by forming a double hydrogen-bonded complex with nitrostyrene(proton acceptor),where the charge transfer between the donor and acceptor increases the eletrophilicity of β-C atom of the nitrostyrene,favoring the nucleophilic attack of the sulfur ylide.
Thiourea;Nitroolefin;Sulfur ylide;Asymmetric Michael addition;Density functional theory
10.3866/PKU.WHXB201201112
O641
∗Corresponding author.Email:zhangdj@sdu.edu.cn;Tel:+86-531-88365833.
The project was supported by the National Natural Science Foundation of China(20873076)and Specialized Research Fund for the Doctoral Program of Higher Education,China(200804220009).
国家自然科学基金(20873076)和教育部博士点基金(200804220009)资助项目
猜你喜欢
化工技术与开发(2022年11期)2022-11-29
北京航空航天大学学报(2022年5期)2022-06-06
化工管理(2021年7期)2021-05-13
科学与财富(2021年33期)2021-05-10
农药科学与管理(2019年8期)2019-11-23
今日农业(2019年11期)2019-08-13
电脑知识与技术(2018年3期)2018-03-21
价值工程(2017年31期)2018-01-17
中国司法鉴定(2017年5期)2017-10-11
哈尔滨理工大学学报(2017年1期)2017-04-08
