
制备方法对CuO-CeO2/ZrO2催化CO氧化性能的影响
2012-11-13 05:50杨志强毛东森杨超杰郭强胜卢冠忠
无机化学学报 2012年7期
杨志强 毛东森 杨超杰 郭强胜 卢冠忠
(上海应用技术学院化学与环境工程学院应用催化研究所,上海 201418)
制备方法对CuO-CeO2/ZrO2催化CO氧化性能的影响
杨志强 毛东森*杨超杰 郭强胜 卢冠忠
(上海应用技术学院化学与环境工程学院应用催化研究所,上海 201418)
以ZrO2为载体、采用不同的浸渍次序制备了3种CuO-CeO2/ZrO2催化剂并在不同的温度(500,650和800℃)下进行焙烧,利用X射线衍射(XRD)、程序升温还原(H2-TPR和CO-TPR)及CO程序升温脱附(CO-TPD)技术对所制备的催化剂进行了表征,并采用色谱流动法考察了其催化CO低温氧化反应性能。结果表明,当焙烧温度为650℃时,3种催化剂的CO催化氧化活性均最佳,且三者的催化活性大小顺序为:CuO/CeO2/ZrO2>CuO-CeO2/ZrO2>CeO2/CuO/ZrO2。结合催化剂的表征和活性测试结果,我们认为高分散的CuO是CO的吸附中心,有利于CO的低温氧化反应,而大颗粒的CuO几乎对CO没有吸附作用,不利于CO的低温氧化反应。在3种催化剂中,CuO/CeO2/ZrO2催化剂具有最佳的低温还原特性和最大的CO2脱附峰面积,相应地具有最佳的催化氧化活性。
浸渍法;CuO-CeO2/ZrO2;催化活性;CO氧化
CO的低温消除在许多方面都有重要的使用价值,如用于汽车尾气净化处理、燃料电池中微量CO的消除、CO中毒防护以及密闭系统中微量CO的脱除等。因此,实现CO在较低温度下转化为CO2已经成为催化研究的热点问题之一[1-2]。由于贵金属催化剂存在价格昂贵且易发生硫中毒等的缺点,CuO/ CeO2催化剂受到国内外同行的广泛关注[1-4]。研究表明,CuO/CeO2催化剂的CO氧化活性远高于单一组分CuO或CeO2[5-6],这主要归因于CeO2能够提高CuO在其表面的分散性并对活性组分CuO起到一个“氧缓冲器”的作用。近年来,CuO/CeO2-ZrO2三元复合氧化物催化剂因比CuO/CeO2催化剂具有更高的热稳定性[7-9]和更长的使用寿命[9]而得到了广泛的研究[7-22]。
CuO/CeO2-ZrO2催化CO氧化反应的性能与其制备方法密切相关。CuO/CeO2-ZrO2催化剂的制备方法主要有浸渍法[7,9-18]、柠檬酸溶胶-凝胶法[8,18-19]和表面活性剂模板法[20]等。其中,浸渍法因具有制备简单、活性组分利用率高和催化活性好等的优点而被广泛采用。目前,浸渍法制备CuO/CeO2-ZrO2催化剂绝大多数都是先制得CeO2-ZrO2,然后再采用浸渍法将CuO负载在CeO2-ZrO2的表面,而以ZrO2为载体来制备CuO-CeO2/ZrO2催化剂则鲜有报道[22]。此外,浸渍法所制备催化剂的性能与活性组分的浸渍次序紧密相关。靳广洲等[23]发现Ce与Cu同时浸渍比先浸Ce再浸Cu所制备的CuO-CeO2/Al2O3催化剂具有更好的催化CO氧化活性。
本工作以ZrO2为载体,采用顺序浸渍法和共浸渍法制备了3种负载型CuO-CeO2/ZrO2催化剂,即CuO负载于CeO2型 (CuO/CeO2/ZrO2)、CuO与CeO2共浸渍型 (CuO-CeO2/ZrO2)和CeO2负载于CuO型(CeO2/CuO/ZrO2)。利用XRD、H2-TPR、CO-TPR和COTPD等测试手段对其物化性质进行了表征,并以CO氧化为探针反应考察了不同浸渍方法所制备的CuO-CeO2/ZrO2催化剂对CO的催化氧化性能。
1 实验部分
1.1 催化剂的制备
采用二次顺序浸渍法制备 CuO/CeO2/ZrO2和CeO2/CuO/ZrO2催化剂,其具体步骤如下:将6.936 1 g的Ce(NO3)3·6H2O溶解于去离子水中,缓慢加入3.601 1 g的商品ZrO2粉末(SBET=7.46 m2·g-1,平均孔径为17.5 nm),磁力搅拌下浸渍4 h后加热烘干至粘稠状,再放入微波炉中快速干燥。然后在空气气氛中于一定温度(500、650或800℃)下焙烧4 h,即制得CeO2/ZrO2前驱体。再以CeO2/ZrO2为载体,加入含2.572 6 g Cu(NO3)2·3H2O的水溶液中进行浸渍,其操作步骤同上,焙烧温度相同,即制得CuO/CeO2/ ZrO2催化剂。ZrO2载体上浸渍硝酸铜和硝酸铈溶液的顺序相反,则可制得CeO2/CuO/ZrO2催化剂。
采用共浸渍法制备CuO-CeO2/ZrO2催化剂:将3.601 1 g的ZrO2粉末缓慢倒入含6.936 1 gCu(NO3)2· 3H2O和2.572 6 g Ce(NO3)3·6H2O的混合水溶液中,其余操作步骤同上。
上述催化剂中CuO和CeO2的总质量百分含量为50%,CuO和CeO2的物质的量的比均为2∶3。所用试剂均为分析纯,由国药集团化学试剂有限公司生产。
1.2 催化剂的表征
采用荷兰Philips公司生产的XPert PRO型X射线衍射仪进行物相分析,以Cu Kα作辐射源(λ=0.154 18 nm),电压和电流分别为40 kV和40 mA,扫描速度为1.0°·min-1,步长为0.02°。根据X-射线衍射线宽法并利用Scherrer公式计算粒子的平均粒径。
H2程序升温还原(H2-TPR)实验在常压微型石英管反应器上进行,试样用量为50 mg,以10%H2-90% N2(体积百分数)的混合气为还原气,升温速率为10℃·min-1,气体流量为50 mL·min-1。还原过程中所生成的水汽用5A分子筛吸收,升温过程中H2的消耗量用浙江温岭福立公司生产的GC9750型气相色谱仪的热导池检测器(TCD)进行检测,检测器的温度为60℃。CO程序升温还原(CO-TPR)与H2-TPR相似,只是还原气为5%CO/He的混合气,且尾气中的CO2用瑞士Balzers公司生产的Omnistar-200型质谱仪跟踪测定。
CO程序升温脱附实验 (CO-TPD)采用瑞士Balzers公司生产的Omnistar-200型质谱仪为检测器,控制m/z为28(CO)和44(CO2)。将0.1 g新鲜催化剂 (40-80目)装入微型石英管反应器中,在He (40 mL·min-1)气氛下以20℃·min-1的速率升温至500℃并维持30 min,然后降至30℃,切换吸附气体CO(40mL·min-1),吸附30min后再切换回He吹扫至无脱附信号,然后以15℃·min-1的速率升温至500℃进行CO脱附实验。
1.3 催化剂反应性能的评价
CO氧化反应在常压微型固定床流动反应装置上进行。将0.2 g催化剂颗粒 (40~60目,250~420 μm)用0.6 g粒度相同的石英砂均匀稀释后,装入下端塞有玻璃棉的内径为6 mm的不锈钢反应器中。在温度为200℃下用高纯N2气(流速为50 mL· min-1)吹扫30min,降至一定温度后切换为反应混合气(体积分数为4%CO、9.85%O2,其余为N2),流速为45mL·min-1。反应前后的气体组成用浙江温岭福立公司生产的GC9790A型气相色谱仪进行在线分析,氢气为载气,TCD检测器,进样口和检测器的温度均为110℃,色谱柱为2 m×3mm的TDX-01碳分子筛不锈钢填充柱。柱温采用一阶程序升温,初始温度40℃,保持5 min后以30℃·min-1的速率升温至130℃,然后再保持5min。在不同温度下测定CO的反应转化情况,直到CO全部转化为CO2为止,停止测定。
2 结果与讨论
2.1 CO催化氧化活性
图1是不同焙烧温度下所制备的3种催化剂对CO氧化反应的结果,其CO完全转化为CO2时的反应温度(T100)数据如表1所示。由图1和表1可以看出,在同一焙烧温度下,3种催化剂的催化活性大小顺序为:CuO/CeO2/ZrO2>CuO-CeO2/ZrO2>CeO2/CuO/ ZrO2,即采用两次浸渍法制备的催化剂,当CuO处于最外层时对CO的催化活性最好。对于不同焙烧温度下制备的催化剂,当焙烧温度为650℃时3种催化剂均具有最佳的催化活性,但与500℃焙烧时的活性相差较小,而经800℃焙烧后,催化剂的活性下降较为明显。该结果与Wang等[19]采用柠檬酸溶胶-凝胶一步法所制备的CuO-CeO2-ZrO2催化剂的活性相一致。
此外,从图1和表1可以发现,CuO-CeO2/ZrO2和CeO2/CuO/ZrO2的催化活性比较接近,但与CuO/ CeO2/ZrO2的催化活性差别比较大,说明催化剂中CuO负载于ZrO2表面并处于CeO2的内层和CuO与CeO2同时负载于ZrO2表面,其催化氧化CO的活性没有发生大的变化,但均低于CuO/CeO2/ZrO2催化剂的活性。也就是说,当CuO处于催化剂的最外层时,其催化氧化CO的活性最高。该结果与靳广洲等[23]的结果不同,他们发现Ce与Cu同时浸渍比先浸Ce再浸Cu所制备的CuO-CeO2/Al2O3催化剂具有更好的催化CO氧化活性。
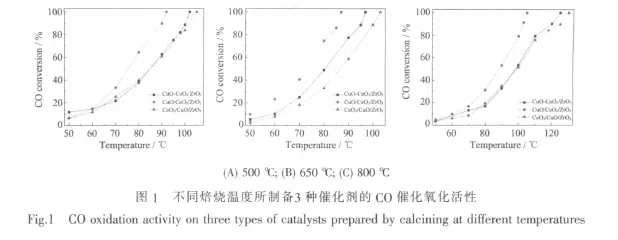

表1 不同焙烧温度所制备3种催化剂的CO催化氧化活性Table 1 CO oxidation activity on three types of catalysts prepared by calcining at different temperatures(T C)
2.2 催化剂的XRD分析
图2为载体ZrO2及不同焙烧温度下所制备3种催化剂的XRD图谱。由此可见,ZrO2在28.2°和31.5°处出现两个较强的衍射峰,表明其以单斜相存在[24]。所有催化剂除出现ZrO2衍射峰外,均在2θ= 28.68°、33.21°、47.69°和56.32°处出现了晶相CeO2的特征衍射峰[25],而且在同一焙烧温度下,CuO/ CeO2/ZrO2中CeO2衍射峰的强度最强,而CeO2/CuO/ ZrO2和CuO-CeO2/ZrO2中CeO2衍射峰的强度相近且较弱;对于同一种催化剂,随焙烧温度的升高,其CeO2衍射峰的强度逐渐增强,尤其是当焙烧温度从650℃升高至800℃时,CeO2衍射峰增强的幅度较大。
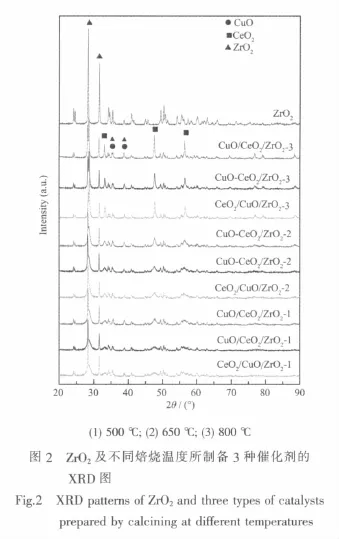
利用Scherrer公式计算了各个催化剂中CeO2的平均粒径,结果如表2所示。从表2中的数据可以看出,当焙烧温度从500℃升高到650℃时,催化剂中CeO2平均粒径的增幅不大,但焙烧温度从650℃进一步升高到800℃时,CeO2平均粒径的增幅达到3倍以上,说明3种催化剂在800℃的高温下焙烧后,其CeO2晶粒均开始快速长大。另外,对于3种不同的催化剂,CuO/CeO2/ZrO2中CeO2晶粒大小随焙烧温度升高而增大的幅度要明显大于 CeO2/CuO/ ZrO2和CuO-CeO2/ZrO2催化剂中CeO2晶粒的增大。这是因为ZrO2作为载体,因其比表面积较小(仅不到8m2·g-1),能够承担CeO2颗粒在其表面分散的能力有限,CeO2颗粒经高温焙烧时很容易生长变大。然而,当CuO和CeO2同时分散于ZrO2颗粒表面时,CeO2颗粒由于受到周围CuO颗粒的 “隔离”作用,抑制了其快速的生长,因此,CuO-CeO2/ZrO2中CeO2颗粒的平均粒径小于CuO/CeO2/ZrO2中CeO2晶粒的粒径。其实,在我们前期研究中已经发现CuO对CeO2颗粒增长有显著的抑制作用[21,26]。出于同样的原因,在ZrO2表面先负载一层CuO后再负载CeO2得到的CeO2/CuO/ZrO2催化剂,其CeO2晶粒的粒径也明显小于 CuO/CeO2/ZrO2催化剂中的CeO2。此外,由于晶相CuO的最强特征衍射峰(2θ= 35.5°和38.7°)与ZrO2的衍射峰重叠较为严重,故无法准确测定其晶粒大小。但由图2可见,不管随催化剂浸渍次序的变化或是焙烧温度的变化,上述衍射峰强度的变化都不明显,这可能是由于CeO2或ZrO2对CuO的烧结均具有较好的抑制作用所致。

表2 不同焙烧温度所制备3种催化剂中CeO2的晶粒大小Table 2 Crystallite size(D)of CeO2 in the three types of catalysts prepared by calcining at different temperatures
2.3 催化剂的H2-TPR分析
图3是不同焙烧温度下所制备3种催化剂的H2-TPR曲线。由此可见,所有样品在125~250℃之间出现部分重叠的宽峰。为此,利用Gaussian法对其进行分峰处理,结果如图中的虚线所示。可见,所有样品均出现2~3个还原峰,按温度由低到高分别被标记为 α、β和 γ峰。根据我们前期研究的结果[14-15],我们认为图中的α峰应归属于催化剂表面与CeO2紧密接触的高分散CuO的还原,β峰应归属于催化剂表面较小颗粒CuO(XRD无法检测)的还原,γ峰应归属于较大颗粒的CuO(XRD可以检测)的还原。各还原峰的峰顶温度(Tmax)及峰面积(S)列于表3中。
由表3可见,对于不同焙烧温度所制备的催化剂,当焙烧温度为650℃时,3种催化剂的α还原峰的温度均最低,说明在这一焙烧温度下CuO颗粒的分散性均最好,其中CuO/CeO2/ZrO2催化剂的α峰还原温度最低,表明CuO颗粒在CeO2载体表面的分散度最高。其次是CuO-CeO2/ZrO2催化剂,由于CuO和CeO2同时被引入至ZrO2的表面,使得CuO同时在CeO2和ZrO2表面分散。但由于CuO在ZrO2表面的分散性不如在CeO2表面好[16],故CuO-CeO2/ ZrO2催化剂中CuO的分散度低于CuO/CeO2/ZrO2催化剂。对于CeO2/CuO/ZrO2催化剂,由于CuO浸渍于ZrO2的表面,其分散性最差,因而α峰的温度最高。此外,当焙烧温度从650℃升高至800℃时,CuO颗粒发生较大程度的烧结,α峰的峰面积均明显减小且还原温度也显著升高。而且,α峰的峰面积随CuO/CeO2/ZrO2、CuO-CeO2/ZrO2和 CeO2/CuO/ZrO2的顺序逐渐减小,直至分不出α峰。
从表 3还可以看出,当焙烧温度为 500和650℃时,β峰和γ峰的Tmax随焙烧温度的升高而升高,但从同一焙烧温度来看,两还原峰的Tmax均依CuO/CeO2/ZrO2、CuO-CeO2/ZrO2和 CeO2/CuO/ZrO2的次序逐渐升高,说明CuO颗粒大小不仅与焙烧温度有关也与制备方法有关。然而经800℃高温焙烧后,β峰和 γ峰的 Tmax和面积却均依 CuO/CeO2/ZrO2、CuO-CeO2/ZrO2和CeO2/CuO/ZrO2的次序逐渐降低,这可能是因为CuO存在于CuO/CeO2/ZrO2催化剂的最外层,无论是较小颗粒或较大颗粒的CuO都可以被H2还原,因此H2-TPR高温还原峰是催化剂中大颗粒CuO的真实反映。而在CeO2/CuO/ZrO2催化剂中,由于CuO被CeO2包裹,特别是大颗粒的CuO被CeO2颗粒紧密包裹,CuO无法被H2充分还原,因此其H2-TPR的高温还原峰不仅峰面积较小而且还原峰的温度较低。
将上述不同催化剂的H2-TPR表征结果与其催化活性进行关联可以发现,其催化CO氧化反应的活性主要与α还原峰相关,即α还原峰的温度越低,其催化活性越高。该结果与Li等[28]的结果基本一致,他们在比较CuO/TiO2、CuO/SnO2和CuO/TiO2-SnO2催化剂的性能时也发现,其催化CO氧化反应的活性与α还原峰相关,α还原峰的温度越低、面积越大,催化剂的活性越高。由此可见,催化剂表面高度分散的CuO物种是其催化CO氧化反应的主要活性中心。
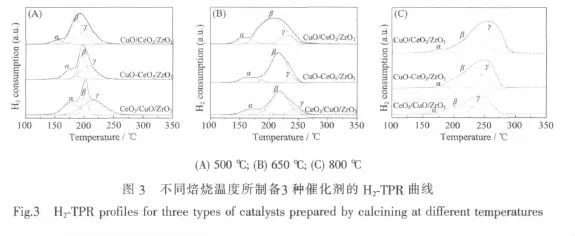
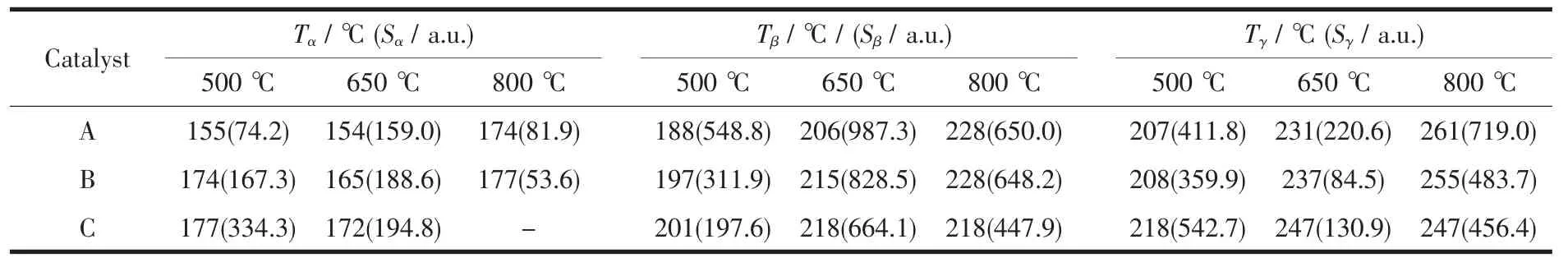
表3 不同焙烧温度所制备3种催化剂的还原峰的温度(T max)和面积(S)Table 3 Temperatures and areas(S)of reduction peaks for the three types of catalysts prepared by calcining at different tem peratures
2.4 催化剂的CO-TPR表征
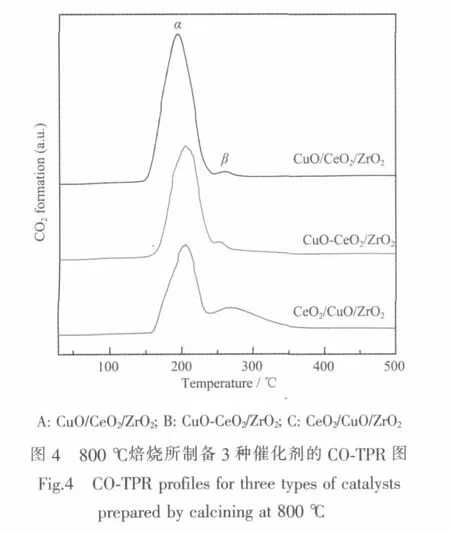
图4是800℃焙烧所制备3种催化剂的COTPR图。可以发现,所有催化剂在200℃左右都出现了一个较大的低温还原峰(α),而在260℃左右出现一个较小的高温还原峰(β)。根据文献[29],α峰对应易还原Cu物种的还原,β峰对应孤立Cu物种的还原。由3种催化剂的CO2脱附峰的温度和面积(表4)可以看出,CuO/CeO2/ZrO2催化剂的α峰温度最低,CO2脱附量最大,CeO2/CuO/ZrO2催化剂的CO2脱附峰的温度与CuO-CeO2/ZrO2相近,但CO2的脱附量最小。表明CuO/CeO2/ZrO2催化剂具有与CO在低温下反应生成CO2的活性中心数目最多,CuO-CeO2/ZrO2次之,CeO2/CuO/ZrO2最少,这与它们的催化活性(表1)相一致。

表4 焙烧温度为800℃时所制备3种催化剂的CO2脱附峰的温度(T max)和面积(S)Table 4 Tem peratures and areas of CO2 desorption peaks for three types of catalysts prepared by calcining at 800℃
2.5 催化剂的CO-TPD表征
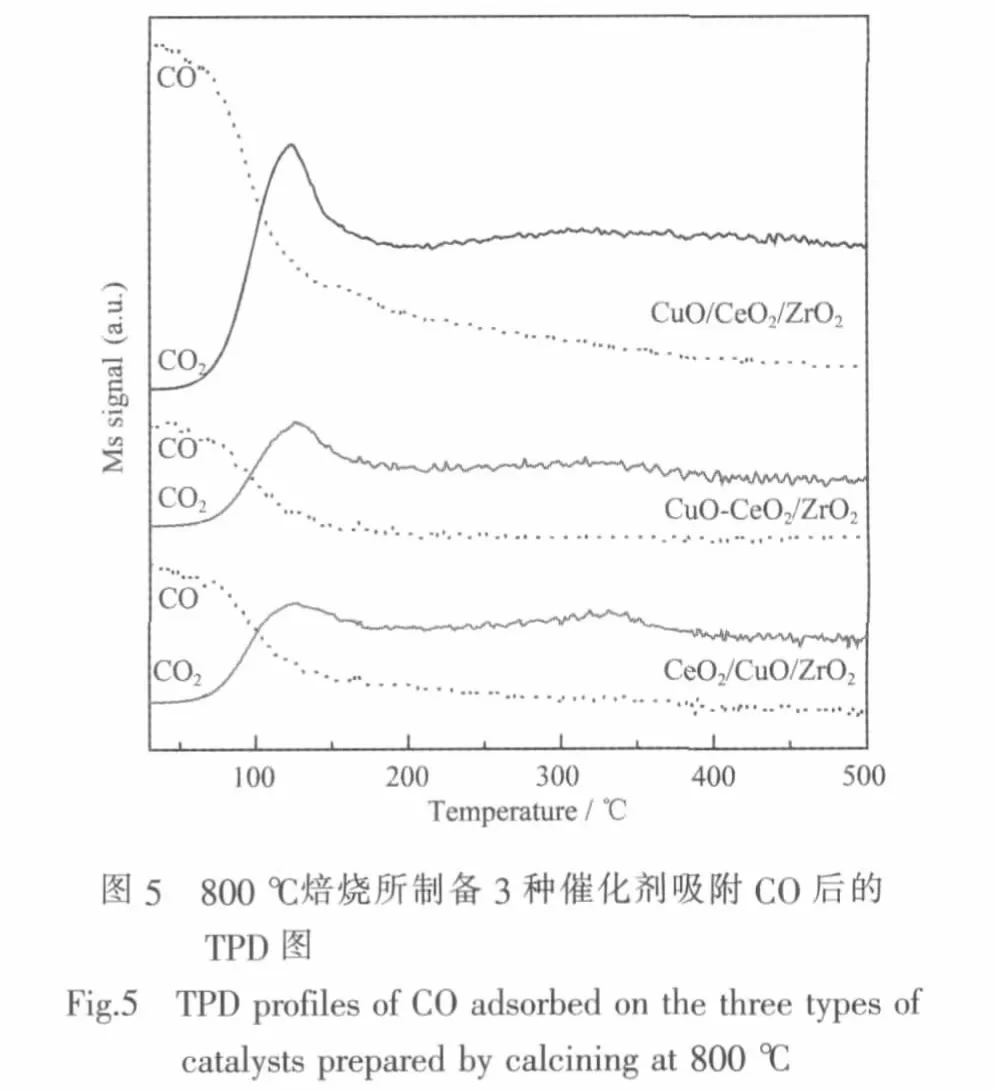
图5为800℃焙烧的3种催化剂吸附CO后的TPD图。图中的虚线表示CO的质谱信号(m/z=28),实线表示CO2的信号(m/z=44)。可以看出,3种催化剂具有相似的CO2脱附曲线,其中在60~200℃区间有一个大的CO2脱附峰,峰顶温度在124℃左右。在200~500℃区间有一个非常弱的脱附峰,峰顶温度在310℃左右。其中,CuO/CeO2/ZrO2吸附CO的能力最强,CO2的脱附量最大,CuO-CeO2/ZrO2与CeO2/CuO/ZrO2相近。Luo[30]等发现在纯的CuO或CeO2颗粒表面上几乎没有任何的 CO吸附,Avgouropoulos等[31]发现纯的CuO表面会产生一个峰顶温度约为100℃的很弱的CO2拖尾峰;而纯的CeO2表面在此温度范围几乎看不到CO2脱附峰。据此可以推测,容易与CO反应的晶格氧来自于CeO2表面上高分散的或较小颗粒的CuO,它们也是催化CO氧化的活性中心。
3 结 论
采用不同的浸渍方式制备了3种不同的CuOCeO2/ZrO2催化剂,当焙烧温度为650℃时,催化剂对CO氧化反应的活性均最佳,且三者的催化活性大小依次为:CuO/CeO2/ZrO2>CuO-CeO2/ZrO2>CeO2/ CuO/ZrO2。结合XRD、H2-TPR、CO-TPR、CO-TPD和活性测试结果,我们认为高分散的CuO是CO的吸附中心,有利于CO的低温氧化反应,而较大颗粒的CuO几乎对CO没有吸附作用,不利于CO的低温氧化反应。3种催化剂中,CuO/CeO2/ZrO2具有最佳的低温还原特性和最大的CO2脱附峰面积,相应地具有最佳的催化氧化活性。同时也说明,活性组分CuO负载于催化剂的外层比分散于催化剂的内层具有更优的催化活性和更佳的低温还原特性。
[1]ZHENG Xiu-Cheng(郑修成),WANG Xiang-Yu(王向宇),YU Li-Hua(于丽华),et al.Prog.Chem.(Huaxue Jinzhan), 2006,18(2/3):159-167
[2]LIANGFei-Xue(梁飞雪),ZHUHua-Qing(朱华青),QINZhang-Feng(秦张峰),et al.Prog.Chem.(Huaxue Jinzhan),2008, 20(10):1453-1464
[3]Luo M F,Ma JM,Lu JQ,etal.J.Catal.,2007,246(1):52-59
[4]MAO Dong-Sen(毛东森),TAO Li-Hua(陶丽华),WANG Qian(王倩),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2010,26(3):447-452
[5]Luo M F,Yan Z L,Jin L Y.J.Mol.Catal.A,2006,260(1/2): 157-162
[6]Radhakrishnan R,Willigan R R,Dardas Z,et al.AIChE J., 2006,52(5):1888-1894
[7]Wang SP,Zheng X C,Wang X Y,et al.Catal.Lett.,2005, 105(3/4):163-168
[8]Wang SP,Wang X Y,Huang J,etal.Catal.Commun.,2007, 8(3):231-236
[9]Wang S P,Zhang T Y,Su Y,et al.Catal.Lett.,2008,121 (1/2):70-76
[10]Luo M F,Zheng X M.Acta Chem.Scand.,1998,52(10): 1183-1187
[11]JIANG Xiao-Yuan(蒋晓原),ZHOU Ren-Xian(周仁贤), CHEN Yu(陈煜),et al.J.Zhejiang Univ.:Sci.Ed.(Zhejiang Daxue Xuebao:Lixueban),2001,28(6):653-658
[12]Chen H L,Zhu H Y,Wu Y,et al.J.Mol.Catal.A,2006, 255(1/2):254-259
[13]Wang SP,Wang X Y,Zheng X C,eta.React.Kinet.Catal. Lett.,2006,89(1):37-44
[14]YANG Zhi-Qiang(杨志强),MAO Dong-Sen(毛东森),ZHU Hui-Lin(朱慧琳),et al.Chinese J.Catal.(Cuihua Xuebao), 2009,30(10):997-1000
[15]YANG Zhi-Qiang(杨志强),MAO Dong-Sen(毛东森),GUO Qiang-Sheng(郭强胜),et al.Acta Phys.-Chim.Sin.(Wuli Huaxue Xuebao),2010,26(12):3278-3284
[16]Ayastuy JL,Gurbani A,González-Marcos M P,et al.Appl. Catal.A,2010,387(1/2):119-128
[17]Zhu J,Zhang L L,Deng Y,et al.Appl.Catal.B,2010,96 (3/4):449-457
[18]HONG Qing-Hong(洪庆红),SONG Yu-Peng(宋宇鹏),JIA Ai-Ping(贾爱平),et al.J.Mol.Catal.(China)(Fenzi Cuihua),2008,22(5):29-433
[19]Wang E G,Chen S Y.J.Rare Earths,2002,20(5):533-537
[20]Cao JL,Wang Y,Zhang TY,etal.Appl.Catal.B,2008,78 (1/2):120-128
[21]YANG Zhi-Qiang(杨志强),MAO Dong-Sen(毛东森),WU Ren-Chun(吴仁春),et al.Acta Phys.-Chim.Sin.(Wuli Huaxue Xuebao),2011,27(5):1163-1168
[22]Águila G,Gracia F,Araya P.Appl.Catal.A,2008,343(1/2): 16-24
[23]JIN Guang-Zhou(靳广洲),YIN Hui-Ling(殷慧玲).J.Univ. Pet.,China:Nat.Sci.Ed.(Shiyou Daxue Xuebao:Ziran Kexue Ban),1997,21(2):76-78
[24]ZHANG Jing(张静),YANSong(阎松).Chinese J.Petrochem. Univ.(Shiyou Huagong Gaodeng Xuexiao Xuebao),2011,24 (1):30-33
[25]YANG Zhi-Qiang(杨志强),MAO Dong-Sen(毛东森),ZHU Hui-Lin(朱慧琳),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2009,25(5):812-817
[26]WANG Qian(王倩),MAO Dong-Sen(毛东森),FANG Zhen-Ni(方珍妮),et al.Chinese J.Inorg.Chem.(Wuji Huaxue Xuebao),2010,26(9):1639-1645
[27]Martínez-Arias A,Fernández-García M,Gálvez O,et al.J. Catal.,2000,195(1):207-216
[28]Li K R,Wang Y J,Wang S R,et al.J.Nat.Gas Chem., 2009,18(4):449-452
[29]Gurbani A,Ayastuy JL,González-Marcos M P,et al.Int.J. Hydrogen Energy,2009,34(1):547-553
[30]Luo M F,Zhong Y J,Yuan X X,et al.Appl.Catal.A, 1997,162(1/2):121-131
[31]Avgouropoulos G,Ioannides T.J.Mol.Catal.A,2008,296 (1/2):47-53
Effect of Preparation M ethod on Catalytic Performance of CuO-CeO2/ZrO2for CO Oxidation
YANG Zhi-Qiang MAO Dong-Sen*YANG Chao-Jie GUO Qiang-Sheng LU Guan-Zhong
(Research Institute of Applied Catalysis,School of Chemical and Environmental Engineering,Shanghai Institute of Technology,Shanghai201418,China)
Three typesof CuO-CeO2/ZrO2catalystswere prepared with ZrO2as the supportby different impregnation sequences,and were calcined atdifferent temperatures(500,650 and 800℃).These catalystswere characterized by X-ray diffraction (XRD),temperature-programmed reduction by H2or CO (H2-TPR,CO-TPR),and temperatureprogrammed desorption of CO (TPD)techniques.Their catalytic activities for low temperature CO oxidation were investigated using amicroreactor-gas chromatograph system.The results indicate that the catalytic activity for the catalysts calcined at650℃is the highestand is in the following order:CuO/CeO2/ZrO2>CuO-CeO2/ZrO2>CeO2/CuO/ ZrO2.Based on the results of characterizations and catalytic activity measurements,we propose that the welldispersed CuO is the center for adsorption of CO and is favorable for low-temperature CO oxidation,and the bulk CuO contributes little to the catalytic activity because of its inability to adsorb CO.Among the three types of catalysts,CuO/CeO2/ZrO2has better low-temperature redox property,larger CO2desorption peak area and higher catalytic activity for CO oxidation.
impregnationmethod;CuO-CeO2/ZrO2;catalytic activity;CO oxidation
O643.3;O614.33+2;O614.41+2
A
1001-4861(2012)07-1353-07
2011-12-05。收修改稿日期:2011-03-15。
上海市教委曙光跟踪计划(10GG23)和上海市教委重点学科建设(J51503)资助项目。
*通讯联系人。E-mail:dsmao@sit.edu.cn;Tel:021-60877221
猜你喜欢
真空与低温(2022年6期)2023-01-06
真空与低温(2022年5期)2022-10-13
云南化工(2020年11期)2021-01-14
石油石化绿色低碳(2019年6期)2019-02-13
劳动保护(2018年8期)2018-09-12
中国资源综合利用(2017年4期)2018-01-22
浙江大学学报(工学版)(2016年11期)2016-06-05
Coco薇(2016年2期)2016-03-22
中国资源综合利用(2016年4期)2016-01-22
河北科技大学学报(2015年5期)2015-03-11
