
氨的氢键数的分子动力学模拟研究
2012-10-12 09:06:14黎多来周昌林李东凯孙振范冯华杰
海南师范大学学报(自然科学版) 2012年4期
黎多来,周昌林,李东凯,孙振范,冯华杰
(海南师范大学 化学与化工学院,海南 海口 571158)
测量液体的氢键需要复杂昂贵的设备,一般条件下难以通过实验来研究.而分子动力学(Molecular Dynamics,MD)是研究流体结构的有力工具,据此可对流体的微观本质作深入、系统的研究[1-4].
大气中的氨是最重要的碱性气体之一,对大气化学和生态平衡产生重要影响[5-8].Ricci等[9]对低温213 K和273 K条件下的液体NH3、ND3、NH3/ND3做了一系列中子衍射实验,获得了液体NH3中存在氢键的实验证据.Boese[10]等采用从头计算量子化学和分子动力学模拟研究了氨的氢键.
本文采用分子动力学模拟方法,使用两种不同的控温方式计算了氨在较宽温度和压力范围的氢键数,并与文献值进行比较,验证了所采用的模拟方法的合理性.
1 模拟方法
MD模拟采用TINKER v5.0程序包,OPLS-AA(optimized potentials for liquid simulations,all-atom)全原子力场[11],参数见表1至表3.表1,2,3中,kb为键伸缩的弹力常数,r0为平衡键长,kθ为键角弯曲的弹力常数,θ0为平衡角,σ与ε为势能参数,σ反映原子间的平衡距离,ε反映势能曲线的深度.

表1 OPLS-AA力场中键的伸缩项参数Tab.1 OPLS-AA Bond Stretching Parameters

表2 OPLS-AA力场中键角弯曲项参数Tab.2 OPLS-AA Angle-Bending Parameters

表3 OPLS-AA力场中非键结势能参数Tab.3 OPLS-AA Non-Bonded Parameters
模拟体系包含300个分子.分子间的作用势采用Lennard-Jones势,体系中的长程作用力采用Ewald[13]加和的形式.在模拟过程中,均采用周期性边界条件、Beeman算法求解运动方程.控温方式分别采用Andersen和Berendsen.体系的截断半径为1 nm,时间步长为1 fs.MD模拟在NVT系综进行,先进行200万时间步(2.0 ns)平衡体系,再进行100万时间步(1.0 ns)用来计算氢键数.
2 结果与讨论
在分子模拟中,氢键的定义有能量标准和几何标准两种[13-14].在本文中,所有体系的氢键判断都是采用几何标准.两个氨分子之间,如果同时满足以下三个条件则认为形成氢键:(1)H—N之间的距离小于2.6 Å;(2)N—N之间的距离小于3.6 Å;(3)键角H—N—N小于60°.
氨的平均氢键数是指氨体系的总氢键数除以总分子数.以Andersen和Berendsen两种不同的控温方式得到的结果分别列于表4和表5.比较表4和表5,可以看到Andersen和Berendsen两种不同的控温方式得到的氨的平均氢键数基本一致.氨的平均氢键数从0.65到2.14,这与Ricci的计算结果[9]吻合.从表中可观察到,氢键数随压力的升高而增大,随温度的升高而减小.另一方面,氢键数受温度的影响比受压力的影响更明显.低温时,氢键数都比较大,在203 K和200 MPa时,氢键数都是高达2.14.而高温时,氢键数较小,在473 K和50 MPa时,氢键数仅为0.65和0.66.也就是说,低温时,氨体系的氢键作用远远强于高温时的氢键作用.

表4 氨的平均氢键数(采用Andersen控温)Tab.4 The H-bond numbers per ammonia molecule(Andersen thermostat)
3 结论
本文采用分子动力学模拟方法研究了氨在较宽温度和压力范围的氢键数,结果表明:Andersen和Berendsen两种不同的控温方式得到的氨的平均氢键数基本一致.随着压力的升高,氢键数增大.而随着温度的升高,氢键数减小.另一方面,氢键数受温度的影响大于受压力的影响.
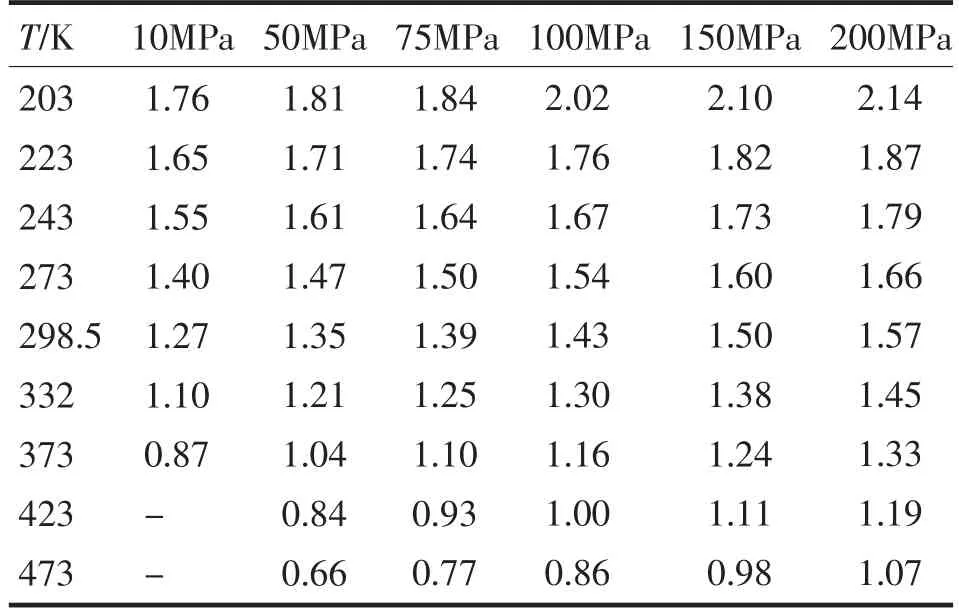
表5 氨的平均氢键数(采用Berendsen控温)Tab.5 The H-bond numbers per ammonia molecule(Berendsen thermostat)
[1]Impey R W,Sprik M,Klein M L.Ionic solvation in nonaqueous solvents:the structure of lithium ion and chloride in methanol,ammonia,and methylamine[J].J Am Chem Soc,1987,109(20):5900-5904.
[2]Diraison M,Martyna G,Tuckerman M.Simulation studies of liquid ammonia by classical ab initio,classical,and path-integral molecular dynamics[J].J Chem Phys,1999,111(3):1096-1103.
[3]Schnabel T,Srivastava A,Vrabec J,et al.Hydrogen bonding of methanol in supercritical CO2:Comparison between1H NMR spectroscopic data and molecular simulation results[J].J Phys Chem B,2007,111(33):9871-9878.
[4]Bauer B A,Patel S.Condensed-phase properties of n-alkyl-amines from molecular dynamics simulations using charge equilibration force fields[J].J Mol Liq,2008,142(1-3):32-40.
[5]Hove L W A,Kooten O,Wijk K J,et al.Physiological effects of long term exposure to low concentrations of SO2and NH3on poplar leaves[J].Physiol Plantarum,1991,82(1):32-40.
[6]Flechard C,Fowler D.Atmospheric ammonia at a moorland site.I:The meteorological control of ambient ammonia concentrations and the influence of local sources[J].Q J Roy Meteor Soc,1998,124(547):733-757.
[7]Robarge W P,Walker J T,Mcculloch R B,et al.Atmo-spheric concentrations of ammonia and ammonium at an agricultural site in the southeast United States[J].Atmos.Environ.,2002,36(10):1661-1674.
[8]Krupa S V.Effects of atmospheric ammonia(NH3)on terrestrial vegetation:a review[J].Environ Pollut,2003,124(2):179-221.
[9]Ricci M A,Nardone M,Ricci F P,et al.Microscopic structure of low temperature liquid ammonia:A neutron diffraction experiment[J].J Chem Phys,1995,102(19):7650-7655.
[10]Boese A D,Chandra A,Martin J M L,et al.From ab initio quantum chemistry to molecular dynamics:The delicate case of hydrogen bonding in ammonia[J].J Chem Phys,2003,119(12):5965-5980.
[11]Rizzo R C,Jorgensen W L.OPLS all-atom model for amines:Resolution of the amine hydration problem[J].J Am Chem Soc,1999,121(20):4827-4836.
[12]Allen M P,Tildesley D J.Computer simulation of liquids[M].Oxford:Claren-don,1987.
[13]Jorgensen W L.Quantum and statistical mechanical studies of liquids.7.Structure and properties of liquid methanol[J].J Am Chem Soc,1980,102(2):543-549.
[14]Jorgensen W L,Ibrahim M.Quantum and statistical mechanical studies of liquids.20.Pressure dependence of hydrogen bonding in liquid methanol[J].J Am Chem Soc,1982,104(2):373-378.
猜你喜欢
中学化学(2024年3期)2024-06-30 15:19:19
航空材料学报(2023年6期)2023-12-18 05:23:50
中学生数理化·八年级物理人教版(2023年6期)2023-05-25 11:59:48
小学生学习指导(小军迷联盟)(2023年3期)2023-03-27 09:22:44
高中数理化(2022年16期)2022-09-14 13:57:06
——《势能》
文化纵横(2022年3期)2022-09-07 11:43:18
中学生数理化·八年级物理人教版(2022年6期)2022-06-05 06:55:40
中国音乐学(2022年1期)2022-05-05 06:48:46
中学生数理化·八年级物理人教版(2021年6期)2021-11-22 07:49:52
青岛大学学报(工程技术版)(2019年2期)2019-09-10 07:22:44
