Aurora激酶抑制剂研究进展*
2012-05-17 09:56袁浩亮张微微陈亚东
药学与临床研究 2012年3期
冷 莹,袁浩亮,张微微,陈亚东,陆 涛
中国药科大学基础部分子设计与药物发现实验室,南京 210009
细胞分裂周期是一个严格调控、高度有序的过程,细胞的有丝分裂对肿瘤细胞的扩增起着关键的调节作用。而有丝分裂的偏差会导致基因组不稳定,这和肿瘤的发生密切相关[1]。Aurora激酶家族是一类能调节中心体和微管功能的丝氨酸/苏氨酸激酶,分为三个亚型:Aurora A、B和C,在细胞有丝分裂的正常进行中发挥至关重要的作用。Aurora激酶异常表达往往会导致细胞在有丝分裂的过程中出现大量的异常现象,很可能引起遗传不稳定和诱发肿瘤的增长[2]。而Aurora激酶独特的药理作用机制以及与恶性肿瘤的关系,使得它成为抗肿瘤药物研究的重要靶点,而其抑制剂也被认为是具有良好开发前景的新型抗肿瘤药物。
1 Aurora激酶的生物学功能及其与肿瘤的关系
Aurora激酶参与了有丝分裂的各项重要事件,包括纺锤体装配、中心体成熟、染色体分离以及胞质分裂等。在正常细胞中,Aurora激酶依靠各种形式的调控手段在特异的时间、空间表达和活化,作用于各种底物分子,从而实现不同的生物学功能[3]。肿瘤的形成是一系列基因突变积累的结果,在各种肿瘤细胞中普遍存在着基因组不稳定的现象,而在许多人类癌细胞中都能观察到Aurora激酶的过表达[4]。1988年首次从结肠癌中克隆出人类的Aurora A和Aurora B基因,同时Aurora A被确定为癌基因。
Aurora A是Aurora激酶家族中最重要的成员,是有丝分裂过程中最主要的调节器。编码基因定位于20q13.2,该区在许多肿瘤中普遍存在扩增,如乳腺癌、结肠癌、卵巢癌和甲状腺癌等[5]。在正常的情况下,Aurora A在有丝分裂过程中的表达和分配是被严格调控的。Aurora A的mRNA在S后期被积累,并在G2/M达到峰值,进入G1期后开始下降。在哺乳动物细胞中,Aurora A定位于中心体,在中心体分离以及细胞分裂过程中发挥重要作用。Aurora A能通过磷酸化Ser625稳定星体关联蛋白(ASAP),这个过程对纺锤体的形成非常重要。此外,Aurora A还能磷酸化中心体蛋白转化酸性含卷曲螺旋蛋白3(TACC3),从而调节微管稳定因子结肠和肝肿瘤高表达基因(ch-TOG),ch-TOG与纺缍体微管的形成有关[6]。并且Aurora A、TACC1和ch-TOG形成的三聚复合物已经在乳腺癌中被证实。Aurora A还会干扰凋亡前体因子,例如p53[7]。Aurora A通过磷酸化Ser215废除p53的结合和反激活功能,它还能磷酸化p53的Ser315位点引起p53不稳定。此外,过表达的Aurora A还能导致基因不稳定,在肿瘤形成之前的主要表现为中心体扩增、染色体四倍性以及过早的姐妹染色单体分离[8]。相反,Aurora A活性的抑制会引起纺锤体瓦解,从而生成一个单一的纺锤体,Aurora A缺陷则会导致有丝分裂延迟以及同源染色体不重合。
Aurora B被称为染色体过客蛋白,编码基因定位在染色体17p13,在原发性结肠癌、乳腺癌以及口腔鳞状上皮癌中都有高表达。Aurora B在有丝分裂的前期至中期定位于着丝粒,进入有丝分裂后期之后逐渐转移到纺锤体中间区域[9]。在有丝分裂过程中,Aurora B能与内部着丝粒蛋白(INCENP)以及生存素在着丝粒上形成复合物。Aurora B会干扰细胞周期以及相关细胞信号转导因子,例如condensin I和哺乳动物雷帕霉素靶蛋白(mTOR)。免疫耗竭的Aurora B能减少对condensin I中Ser10位点的磷酸化,导致condensin I相关的染色体减少大约50%。而在T细胞中,mTOR抑制剂雷帕霉素能降低Aurora B激酶的活性,抑制Aurora B介导的细胞周期进程[10]。研究表明,Aurora B能够磷酸化Mis13的Ser100和Ser109位点,促进着丝点装配的进行。因此抑制Aurora B激酶会引起着丝点装配的抑制,从而导致多核细胞的形成。在有丝分裂过程中,Aurora B能磷酸化Ser10/Ser28位点,从而干扰组蛋白H3,引起异染色质蛋白1(HP1)的解离。此外Aurora B还能干扰细胞信号转导通路。
Aurora C也是一种染色体过客蛋白,和Aurora B有相同的分布。在有丝分裂中,Aurora C在前期和中期定位于着丝粒,后期定位在微管中间区域[11]。免疫共沉淀表明Aurora C能够和Aurora B及survivin发生作用,在体内还能观察到Aurora C和INCENP的联系。虽然目前Aurora C的生物功能还不是很清楚,但是普遍认为Aurora C和Aurora B功能相似,在胞质分裂中发挥相似的作用。Aurora C或者Aurora B的过表达会导致多核细胞的形成。相反,野生型的Aurora C激酶的过表达能够减少由于Aurora B激酶突变诱导的多核细胞的数量。Aurora C在哺乳动物的睾丸中高表达,能够促进精子的产生并对纤毛和鞭毛进行调节。Aurora C在肿瘤中的相关报道较少,但在原发性结肠癌和一些癌细胞系中检测到了过表达的Aurora C。
2 Aurora激酶活性位点的结构特征
Aurora激酶的三个亚型在结构上具有高度保守性,有一个长度在309~403范围内的氨基酸序列,包括N末端区(39~129)、蛋白激酶区以及C末端区(15~20)[12]。 蛋白激酶区具有典型的 bilobal结构,其中较小的N-末端残片包含一个五股反向的β片状结构和两个α螺旋结构 (氨基酸残基127~215)。较大一点的C-末端残片为α螺旋结构(氨基酸残基216~385),含与多肽底物结合有关的活性环。Aurora A和Aurora B在C末端催化区域的氨基酸序列保守性达到71%,这对底物和抑制剂的特异性非常重要。
Aurora激酶抑制剂主要是通过竞争性的作用于疏水性的ATP结合口袋从而发挥抑制作用。目前已经有Aurora A和Aurora B激酶的晶体结构报道,为小分子抑制剂的研究提供了重要的结构信息。晶体结构研究表明,Aurora激酶的活性位点可以划分成四个部分:溶剂暴露前口袋区、铰链区、疏水的后口袋区以及磷酸盐结合区域[13](如图1所示)。ATP结合口袋的进入由一个看门氨基酸残基Leu210控制。溶剂暴露前口袋由氨基酸残基Tyr212、Ala213、Pro214、Leu215、Gly216、Arg220、Lys224、Leu264、Gly265、Ser266、Arg137、Leu139 和 Phe157组成。铰链区由氨基酸残基210–216组成,是氢键网络形成的主要区域,并且大多数小分子抑制剂都是通过和主链上的Glu211以及Ala213形成直接的氢键作用而结合在激酶铰链区上。疏水的后口袋区是一个选择性口袋,存在于大部分激酶中。在Aurora激酶中该位点是由铰链区的Leu210和Glu211、甘氨酸富集环中的Val147,以及激酶中的Ala160和Leu194组成。这个疏水的后口袋是非保守的,主要用于增加亲和力和选择性。高度溶剂暴露的磷酸盐结合区域由氨基酸残基Lys143、Phe144、Lys162、Leu164、Leu178、Glu181、Leu194、Leu208、Arg255、Lys258、Glu260、Asn261、Leu263、Lys271、Ile272、Ala273、Asp274、Phe275 和 Trp277 组成,比溶剂暴露前口袋区大得多。结构分析研究表明,溶剂暴露前口袋区和磷酸盐结合区都在ATP结合口袋的外部,并且高度暴露在溶剂中,因此,可以根据这两个区域的特性来对候选药物进行结构修饰,从而改善其药代动力学性质。
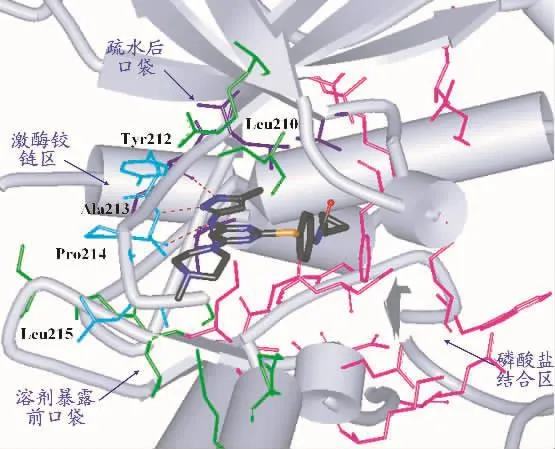
图1 Aurora激酶活性位点结构
3 Aurora激酶抑制剂的研究进展
Aurora激酶作为抗癌药物发展中的一个重要靶标,已经吸引了很多制药公司和科研机构对其抑制剂进行研究和开发。目前已经有大量关于Aurora激酶小分子抑制剂的报道,其中部分化合物已经进入临床研究阶段,如表1所示。
3.1 第一代Aurora激酶抑制剂
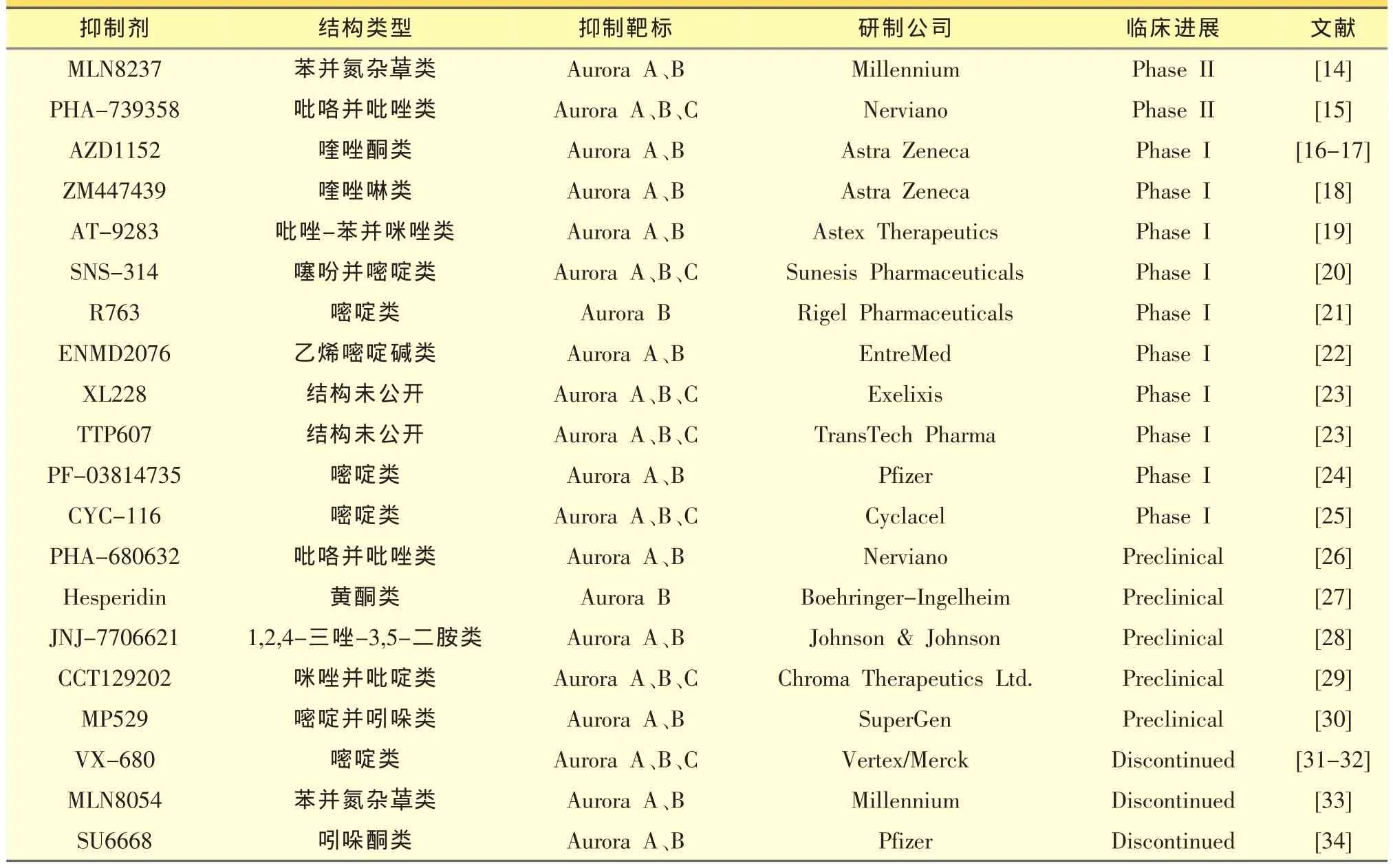
表1 处于临床研究阶段的Aurora激酶抑制剂
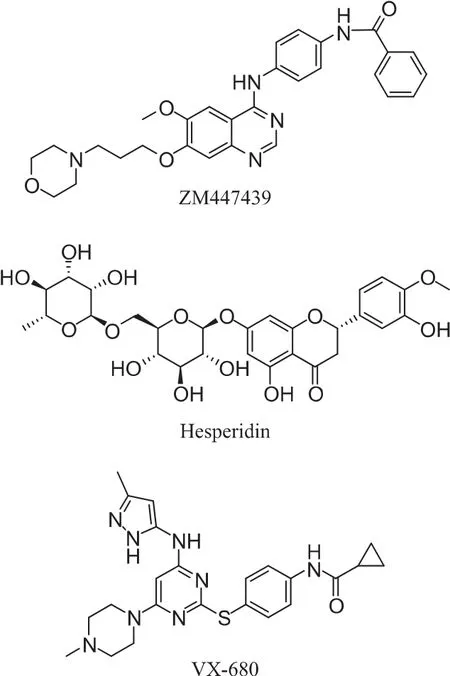
ZM447439(喹唑啉类)是从250,000多个对Aurora A激酶具有抑制活性的化合物中筛选出来的一类喹唑啉衍生物,抑制Aurora A和B的IC50值分别为 110 nmol·L-1和 130 nmol·L-1。 它能调控细胞中的染色体排列、细胞分裂和有丝分裂检查点[18]。ZM447439的介导作用能够减少组蛋白H3的磷酸化,抑制BUBR1、MAD2以及Cenp-E着丝点的定位,主要是对Aurora B产生抑制作用。最新研究发现,ZM447439对血液恶性肿瘤包括急性骨髓(AML)和费城染色体阳性急性淋巴细胞白血病均有抑制活性。在有p53缺陷的细胞中,ZM447439能够增强核内复制,表明p53非依赖性机制可能对ZM447439诱导的四倍性有影响。目前,ZM447439已进入I期临床研究阶段。构效关系研究表明,苯甲酰苯胺是一个疏水口袋结合基团,用小的亲脂性基团如3-氟苯胺、2,3-二氟苯胺和3-氟乙酰苯胺取代苯胺基更有利于终端和疏水结合口袋的结合。而用吡唑环取代连接喹唑啉母核的苯胺基团,能提高Aurora B的特异选择性。
Hesperidin(黄酮类)是 Bohringer公司于 2001年通过高通量筛选的方式得到的一个黄酮类化合物。最初是作为一个广谱的激酶抑制剂,2003年发现了它能通过ATP竞争的方式特异性的抑制Aurora B,IC50值为 259 nmol·L-1[27]。 同时对其他激酶如AMPK、LCK、MKK1、MAPKAP-K1、CHK1 和 PHK 等都能产生交叉反应。Hesperidin能延迟细胞在有丝分裂过程中从中期到后期的转变,并且使细胞在中心体分离之后就过早地退出有丝分裂。药理实验表明,Hesperidin能够定位到着丝点,从而破坏检查点蛋白,并且能诱导胞质分裂以及多倍性。Aurora B/INCENP/Hesperadin晶体复合物的分析表明,Hesperadin的吡喃酮部分能够和Aurora B的催化区相结合,从而对Aurora B起到抑制作用。Hesperidin是第一个被证实的Aurora B抑制剂,但在临床试验上还没有完全被证实。
VX-680/MK0457(嘧啶类)是基于Aurora激酶的ATP结合口袋设计的一个4,6-二氨基嘧啶衍生物,它靶向 ATP结合位点,抑制 Aurora A、B、C的 IC50值分别为 0.6、18、4.6 nmol·L-1[31]。 肿瘤细胞水平的实验表明,VX-680抑制细胞增殖的IC50值为15~113 nmol·L-1,对 AML患者体内获得的白血病细胞的抑制活性IC50值为35~100 nmol·L-1。动物实验研究表明,当VX-680按一天2次腹膜内给药,剂量为 75 mg·kg-1,连续给药 13天,能够成功地减小肿瘤体积[32]。VX-680还能和野生型及突变型的Abl激酶结合,在Bcr-Abl突变体中VX-680抑制细胞增殖的 IC50值在 100到200 nmol·L-1之间。Bcr-Abl的突变体对抗肿瘤药物伊马替尼、尼罗替尼和达沙替尼具有耐药性,因此VX-680可能成为治疗与Abl相关肿瘤的新途径。VX-680在2005年用于I期临床试验,治疗慢性髓细胞白血病(CML)以及T315I Bcr-Abl突变的急性淋巴细胞性白血病(ALL)。但是由于可能引起的QTc延长风险,Merck公司在2007年暂停了已经处于I~Ⅱ期临床的VX-680的注册。
3.2 Aurora激酶的多重抑制剂
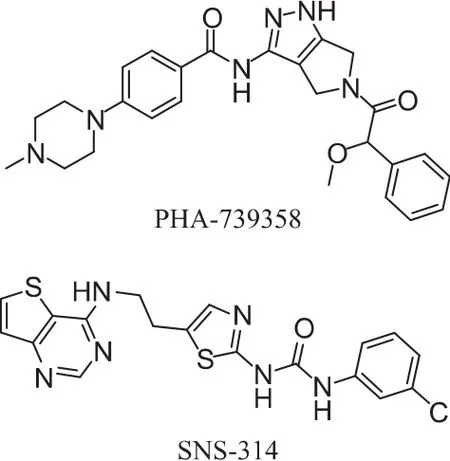
Aurora激酶的多重抑制剂对 Aurora A、B、C均有抑制作用,其中典型的代表药物有PHA-739358和 SNS-314。
PHA-739358(吡咯并吡唑类)是在临床前候选化合物PHA-680632(吡咯并吡唑类)的基础上设计改造而成的,是ATP竞争性抑制剂,其对Aurora A、B、C 抑制活性 IC50值分别为 13、79、61 nmol·L-1[15]。对其他激酶如Abl、Ret、Trk-A抑制活性的IC50值分别 为 25、31、30 nmol·L-1, 它 还 能 抑 制 c-Abl 在Tyr393的自体磷酸化以及Brc-Abl下游信号分子的磷酸化。交叉验证表明,PHA-739358对FGFR-1的抑制作用是Aurora A的四倍。PHA-739358能够诱导细胞多倍性的产生。最近PHA-739358已经进入Ⅱ期临床研究,用于伊马替尼治疗后复发的慢性粒细胞白血病以及靶向c-Abl治疗之后癌细胞转移的前列腺癌。
SNS-314(噻吩并嘧啶类)对 Aurora A、B、C 都有很好的抑制作用,IC50分别为 9、31、3 nmol·L-1,是从高通量筛选的方式得到的先导化合物优化而来,已经处于1期临床研究阶段[20]。在分子和细胞水平,SNS-314能抑制组蛋白H3磷酸化并且中断有丝分裂。SNS-314还能抑制各种人类肿瘤细胞的增殖,例如 HT-29、PC3、HeLa 和 A375,并且 BrdU IC50值在 1.8~9.3 nmol·L-1之间。 SNS-314能减小结肠癌细胞的体积,此外,对黑素瘤、前列腺癌以及肺癌细胞都有很强的抑制活性。该化合物于2007年8月进入I期临床研究,主要用于晚期实体瘤的治疗。
3.3 Aurora A的特异性抑制剂
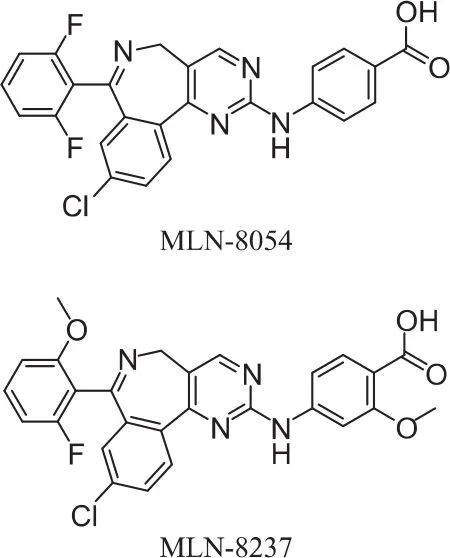
体外酶活性实验表明,Aurora A激酶的选择性抑制剂非常值得关注。MLN8054和MLN8237是ATP竞争性的Aurora激酶可逆抑制剂,两者都对Aurora A具有选择性,并且对Aurora A的敏感性比Aurora B大约高出40倍和200倍。
点评:婚姻的第一毒瘤就是托付心态。何谓托付心态?就是将自己的成功、快乐、幸福的权利交给外界来掌控。抱着这种心态的人,不会有成熟的关系。因为她会有“你应该为我打理好一切”、“你应该成为人上人”的妄念。这种妄念会导致她在风雨面前,不思进取、推卸责任、怨天尤人。正确的心态应该是:自己才是问题最好的解决者。在困难面前,做一个负责人的成年人,与他一起互相支持、共同成长,开创未来。
3.4 Aurora B的特异性抑制剂
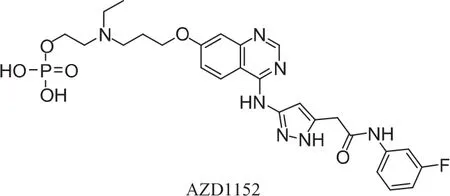
AZD1152(喹唑酮类)是第一个进入临床研究的Aurora B选择性小分子抑制剂,抑制Aurora A和B的 IC50值分别为 1.36 nmol·L-1、0.37 nmol·L-1。 它是乙酰基苯胺取代的吡唑-氨基喹唑啉前体药物,能在人类血浆中快速转化成活性成分AZD1152-羟基喹唑啉吡唑乙酰基苯胺 (HQPA),AZD1152-HQPA对Aurora B有选择性抑制作用[16]。此外,AZD1152对50多种其它类型的丝氨酸/苏氨酸激酶,以及包括FLT3、JAK2、ABL在内的酪氨酸激酶均有抑制活性。
研究表明,AZD1152在体内能够显著抑制肿瘤生长,在一些情况下还能引起显著的肿瘤消退,在用AZD1152处理的动物中,观察到了磷酸化组蛋白H3的减少,4N或更高DNA含量细胞的增加,多核细胞产生以及细胞凋亡的增多,同时也观察到了显著却短暂的骨髓细胞的减少,但在最后一次用药后五天即可恢复[17]。临床前研究表明,AZD1152能显著地抑制淋巴细胞或者血小板的功能,但是没有出现淋巴球减少症和血小板减少症。AZD1152目前已经处于临床I期,用于治疗晚期实体肿瘤,以静脉注射的方式给药。AZD1152能协同增加长春新碱和柔红霉素的抗肿瘤效应。
3.5 多靶点的Aurora激酶抑制剂
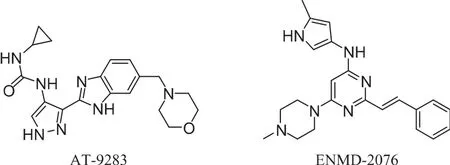
多靶点的Aurora激酶抑制剂不仅能抑制Aurora激酶,还对多个肿瘤治疗靶点有抑制作用,其中典型的代表有AT-9283和ENMD-2076。
AT-9283(吡唑-苯并咪唑类)是Astex公司采用基于药物的碎片发现方法获得的一个多靶点激酶抑制剂,能够抑制酪氨酸以及丝氨酸/苏氨酸激酶, 例如 Aurora A 和 B、JAK-2 和-3、Tyk2、RSK2,体外测得的IC50值都小于10 nmol·L-1[19]。AT-9283对野生型BCR-ABL或者突变的BCR-ABL以及BAF3均有强的抗增殖活性。该化合物的I/II期临床实验主要是用于早期实体瘤和白血病的研究。AT-9283在I期临床治疗实体肿瘤中,通过静脉注射72 h,每3周剂量按比例增加的方式给药,最大耐受剂量为9 mg·m-2·d-1。 同时在2008年的ASCO会议中公布的另一个用于白血病的I期临床研究表明:在AML、ALL、MDS以及CML患者中,连续72 h输注AT-9283,最大耐受剂量为 108 mg·m-2·d-1,到目前为止尚未观察到明显的不良反应。
ENMD-2076(乙烯嘧啶碱类)是一个口服有效的乙烯嘧啶碱类化合物,选择性的抑制Aurora A,对Aurora A和B抑制活性的IC50值分别为14 nmol·L-1和 290 nmol·L-1。 它还能抑制 Src、cKit、FAK、VEGFR2,IC50值分别为 100、40、5、80 nmol·L-1。研究表明,ENMD-2076能使细胞周期停滞在G2/M期,最终导致细胞凋亡[22]。在临床前的动物模型中,ENMD-2076使得组蛋白H3阳性的卵巢癌细胞1A9的比例增加到80%。动物实验研究显示,ENMD-2076能降低VEGFR2活性并且诱导对结肠癌的抗肿瘤作用。这些研究表明,ENMD-2076具有抗血管生成、阻碍细胞周期以及抗增殖活性,这为该化合物用于治疗包括多发性骨髓瘤在内的血液癌提供了支持。ENMD-2076的I期临床研究始于2008年3月,用于治疗实体肿瘤,目前已经制定了治疗血液瘤的I期临床研究计划。
4 Aurora激酶抑制剂的开发策略
由于Aurora激酶亚型在有丝分裂中的作用和功能存在差别,抑制剂究竟是靶向单一的Aurora激酶亚型,还是同时作用于三个亚型以起到最大的抗肿瘤作用,至今还未阐明。为了进一步实现这个目标,从肿瘤生物学角度去开发一些靶向作用于单个Aurora激酶亚型的选择性抑制剂是很有必要的。目前已经有一些Aurora激酶抑制剂进入临床研究阶段,包括非选择性Aurora激酶抑制剂、Aurora A选择性抑制剂、Aurora B/C选择性抑制剂以及多靶点激酶抑制剂等[35]。
近年来已经有少量的对多种激酶都有抑制作用的Aurora激酶抑制剂进入临床研究。这类抑制剂比特异性的靶向Aurora激酶的抑制剂更能增加有效治疗的机会,并且作为不同类型的抗肿瘤药物,它还能降低耐药性的产生。目前已经有多靶点的激酶抑制剂成功地用于癌症治疗的实例,例如舒尼替尼和伊马替尼等。因此,与同时作用于多个靶点的抑制剂相比,特异性靶向Aurora激酶的抑制剂的优势目前仍存在疑问。
联合治疗是目前临床上癌症治疗的重要策略,而Aurora激酶选择性抑制剂和其他的抗肿瘤治疗手段的结合可增强抗肿瘤作用[36]。例如,放射治疗和AZD1152联合使用能增加癌症治疗的敏感性;MK-0457和多西他赛的联合使用在卵巢癌治疗中起到增加抗肿瘤作用的效果。而目前对Aurora激酶抑制剂的联合治疗研究并不是很深入,因此今后在临床试验设计中加入适当的联合治疗药物对肿瘤治疗有非常重要的意义。
5 总结和展望
近年来人们对Aurora激酶作为抗肿瘤药物靶点进行了大量的深入研究,推动了Aurora激酶抑制剂的不断向前发展,使其成为抗肿瘤药物研究的热点之一。本课题组采用基于碎片的药物设计方法得到了一些结构新颖的、具有Aurora抑制活性的化合物,对Aurora激酶抑制剂的研究提供了一些新的思路。Aurora激酶作为一个重要的有丝分裂调节器,从目前的发展趋势来看,Aurora激酶抑制剂的发展前景广阔,很有可能在未来抗肿瘤新药的开发中占据重要的地位。
[1] Pérez de Castro I,de Cárcer G,Malumbres M.A census of mitotic cancer genes new insights into tumor cellbiology and cancertherapy [J].Carcinogenesis,2007,28(5):899-912.
[2] Fu J,Bian M,Jiang Q,et al.Roles of Aurora kinases in mitosis and tumorigenesis[J].Mol Cancer Res,2007,5(1):1-10.
[3] Gautschi O,Heighway J,Mack PC.Aurora kinases as anticancer drug targets[J].Clin Cancer Res,2008,14(6):1639-48.
[4] Mountzios G,Terpos E,Dimopoulos MA.Aurora kinases as targets for cancer therapy[J].Cancer Treat Rev,2008,34(2):175-82.
[5] Lu LY,Wood JL,Ye L,et al.Aurora A is essential for early embryonic development and tumor suppression[J].J Biol Chem,2008,283(46):31785-90.
[6] Cheeseman LP,Booth DG,Hood FE,et al.Aurora A kinase activity is required for localization of TACC3/ch-TOG/clathrin inter-microtubule bridges[J].Commun Integr Biol,2011,4(4):409-12.
[7] Agnese V,Bazan V,Fiorentino FP,et al.The role of Aurora-A inhibitors in cancer therapy[J].Ann Oncol,2007,18(6):47-52.
[8] Hégarat N,Smith E,Nayak G,et al.Aurora A and Aurora B jointly coordinate chromosome segregation and anaphase microtubule dynamics[J].J Cell Biol,2011,195(7):1103-13.
[9] Tanenbaum ME,Medema RH.Localized Aurora B activity spatially controlsnon-kinetochore microtubules during spindle assembly[J].Chromosoma,2011,120(6):599-607.
[10] Portella G,Passaro C,Chieffi P.Aurora B:a new prognostic marker and therapeutic target in cancer[J].Curr Med Chem,2011,18(4):482-96.
[11] Sen S.Aurora-C:the youngest of"the three(Aurora kinase)tenors"of mitotic symphony[J].Cell Cycle,2009,8(19):3076-7.
[12] Yan A,Wang L,Xu S,et al.Aurora-A kinase inhibitor scaffolds and binding modes[J].Drug Discov Today,2011,16(5-6):260-9.
[13] Vulpetti A,Bosotti R.Sequence and structural analysis of kinase ATP pocket residues[J].Farmaco,2004,59(10):759-65.
[14] Carol H,Boehm I,Reynolds CP,et al.Efficacy and pharmacokinetic/pharmacodynamic evaluation of the Aurora kinase A inhibitor MLN8237 against preclinical models of pediatric cancer[J].Cancer Chemother Pharmacol,2011,68(5):1291-304.
[15] Carpinelli P,Ceruti R,Giorgini ML,et al.PHA-739358,a potent inhibitor of Aurora kinases with a selective target inhibition profile relevant to cancer[J].Mol Cancer Ther,2007,6(12 Pt 1):3158-68.
[16] Aihara A,Tanaka S,Yasen M,et al.The selective Aurora B kinase inhibitor AZD1152 as a novel treatment for hepatocellular carcinoma[J].J Hepatol,2010,52(1):63-71.
[17] Lo¨wenberg B,Muus P,Ossenkoppele G,et al.Phase 1/2 study to assess the safety,efficacy,and pharmacokinetics of barasertib (AZD1152)in patients with advanced acute myeloid leukemia[J].Blood,2011,118(23):6030-6.
[18] Li M,Jung A,Ganswindt U,et al.Aurora kinase in-hibitor ZM447439 induces apoptosis via mitochondrial pathways[J].Biochem Pharmacol,2010,79(2):122-9.
[19] Kimura S.AT-9283,a small-molecule multi-targeted kinase inhibitor for the potential treatment of cancer[J].Curr Opin Investig Drugs,2010,11(12):1442-9.
[20] Arbitrario JP,Belmont BJ,Evanchik MJ,et al.SNS-314,a pan-Aurora kinase inhibitor,shows potent anti-tumor activity and dosing flexibility in vivo[J].Cancer Chemother Pharmacol,2010,65(4):707-17.
[21] McLaughlin J,Markovtsov V,Li H,et al.Preclinical characterization of Aurora kinase inhibitor R763/AS703569 identified through an image-based phenotypic screen[J].J Cancer Res Clin Oncol,2010,36(1):99-113.
[22] Zhang S,Farag SS.From cell biology to therapy:ENMD-2076 in the treatment of multiple myeloma[J].Expert Opin Investig Drugs,2011,20(7):1015-28.
[23] Dar AA,Goff LW,Majid S,et al.Aurora kinases’inhibitors-rising stars in cancer therapeutics[J].Mol Cancer Ther,2010,9(2):268-78.
[24] Hook KE,Garza SJ,Lira ME,et al.An integrated genomic approach to identify predictive biomarkers of response to the Aurora kinase inhibitor PF-03814735[J].Mol Cancer Ther,2012,11(3):710-9.
[25] MacCallum D,Melville J,Campbell K,et al.Combination studies with the oral Aurora kinase inhibitor CYC116 and chemotherapeutic agents[C].Proceedings of the 99th AACR Annual Meeting,2008.
[26] Soncini C,Carpinelli P,Gianellini L,et al.PHA-680632,a novel Aurora kinase inhibitor with potent antitumoral activity[J].Clin Cancer Res,2006,12(13):4080-9.
[27] Jetton N,Rothberg KG,Hubbard JG,et al.The cell cycle asa therapeutic targetagainstTrypanosoma brucei:Hesperadin inhibits Aurora kinase-1 and blocks mitotic progression in bloodstream forms[J].Mol Microbiol,2009,72(2):442-58.
[28] Danhier F,Ucakar B,et al.Magotteaux N,active and passive tumor targeting of a novel poorly soluble cyclin dependent kinase inhibitor,JNJ-7706621[J].Int J Pharm,2010,392(1-2):20-8.
[29] Chan F,Sun C,Perumal M,et al.Mechanism of action of the Aurora kinase inhibitor CCT129202 and in vivo quantification of biological activity[J].Mol Cancer Ther,2007,6(12):3147-57.
[30] Joshi-Hangal R,Tang C,Sadikin S,et al.Pharmacokinetics of MP529,a selective Aurora A kinase inhibitor,in a novel subcutaneous delivery system[C].AACR Meeting Abstracts,2008.
[31] Tyler RK,Shpiro N,Marquez R,et al.VX-680 inhibits Aurora A and Aurora B kinase activity in human cells[J].Cell Cycle,2007,6(22):2846-54.
[32] Harrington EA,Bebbington D,Moore J,et al.VX-680,a potent and selective small-molecule inhibitor of the Aurora kinases,suppresses tumor growth in vivo[J].Nat Med,2004,10(3):262-7.
[33] Manfredi MG,Ecsedy JA,Meetze KA,et al.Antitumor activity of MLN8054,an orally active smallmolecule inhibitor of Aurora A kinase[J].Proc Natl Acad Sci USA,2007,104(10):4106-11.
[34] Godl K,Gruss OJ,Eickhoff J,et al.Proteomic characterization of the angiogenesis inhibitor SU6668 reveals multiple impacts on cellular kinase signaling[J].Cancer Res,2005,65(15):6919-26.
[35] Vader G,Lens SM.The Aurora kinase family in cell division and cancer[J].Biochim Biophys Acta,2008,1786(1):60-72.
[36] Kitzen JJ,de Jonge MJ,Verweij J.Aurora kinase inhibitors[J].Crit Rev Oncol Hematol,2010,73(2):99-110.
猜你喜欢
中风与神经疾病杂志(2022年9期)2022-10-19
电气技术(2022年5期)2022-05-23
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
汽车工程师(2021年12期)2022-01-18
第一财经(2019年8期)2019-08-26
科学24小时(2018年1期)2018-01-10
分析化学(2017年12期)2017-12-25
作文·初中版(2017年6期)2017-06-16
现代养生·下半月(2016年6期)2016-10-21