
一种温敏复制缺陷T载体的构建及在鸡白痢沙门氏菌基因敲除中的应用
2012-02-09 00:55郭春林蕾任妮妮姜轲冉袁海霞余旭平
生物工程学报 2012年9期
郭春,林蕾,任妮妮,姜轲冉,袁海霞,余旭平
浙江大学动物科学学院 农业部动物病毒学重点实验室,浙江 杭州 310058
基因敲除是后基因组时代基因功能研究的最有效手段之一,而载体是基因敲除工作的工具和核心[1]。2010年李军华等[2]以pBS246质粒为骨架,在该质粒的两个LoxP序列之间插入正筛选标记基因,并在两个LoxP序列外侧分别插入两组携带“8 碱基”酶切位点的多克隆位点序列(MCS-1和MCS-2) 和负筛选标记基因,成功构建了通用型基因打靶载体 pGT-V1,该载体的构建为有效开展基因打靶提供了新的平台。同年姜娜等[3]通过构建打靶载体pHY-Kan-ssaV,成功敲除了伤寒沙门氏菌S.ty2的ssaV基因,获得伤寒沙门氏菌 ssaV基因缺失株。基因缺失株的构建是研究病原微生物致病机理的重要手段[4],通过构建致病菌的基因缺失株并分析其表型[5],有助于快速了解致病基因功能和致病机制。2007年乔凤等[6]在pUC19质粒的基础上,在其多克隆位点处插入卡那霉素抗性基因,并在该基因两侧添加多个酶切位点,成功构建了pUC19K质粒。作者进一步利用该自杀质粒构建了布鲁氏菌外膜蛋白 Omp25基因突变株,为细菌基因功能研究奠定基础。2011年胡勇等[7]以pMD18-T为载体,利用交错PCR技术构建了伤寒沙门氏菌bcfD基因敲除突变株,为进一步研究 bcfD的基因功能提供有效的技术手段。
基因敲除方法很多,插入复制突变法(Insertion duplication mutation,IDM) 是其中一种简单高效的基因敲除和基因功能研究方法[8-9]。IDM是利用克隆于载体中的DNA片段与基因组中的相应基因同源重组,插入另一拷贝DNA片段和质粒骨架,导致重组菌中相应基因被截断而敲除。IDM可以用于基因功能的鉴定,包括细菌基因的必需性鉴定。2004年Knuth 等[8]采用IDM方法鉴定出鼠伤寒沙门氏菌ATCC14028的 257个必需基因,该方法构建了一个带温度敏感复制子的pIDM1质粒,它在37 ℃时不能复制;利用该载体的 KpnⅠ位点,Knuth等[8]克隆了基因组随机PCR (rPCR) 产物,构建了鼠伤寒沙门氏菌本菌株基因组文库。通过细菌培养温度的改变,筛选出发生了同源重组并插入 pIDM1骨架 (含tet基因) 的重组菌。由于必需基因的IDM敲除菌株不能存活,理论上不能通过培养温度转换而筛选到重组菌 (实际上因非同源重组、突变等原因,仍可能筛到极少量的四环素耐药菌株),而非必需基因的敲除不影响细菌的生存,因此能筛选到大量的重组菌,计算重组率 (Integration rate per cell,IPC) 可以了解基因的必需性[9]。2007年,Klumpp等[10]还进一步测定经pIDM1质粒突变的菌株在巨噬细胞中的存活情况,筛选获得与巨噬细胞内复制相关的致病基因。
pIDM1是一个有效的基因敲除载体,但pIDM1质粒需要经KpnⅠ等单酶切,并需进一步脱磷处理以避免自身环化,操作较繁琐,成功率低;另一方面,PCR产物也需首先经过酶切,而不能进行直接克隆。为此,本实验拟在 pIDM1的基础上,在EcoRⅠ和PstⅠ位点间插入XcmⅠ接头,构建可以直接克隆PCR产物的T载体,进一步应用构建的T载体进行鸡白痢沙门氏菌2个基因的敲除和必需性检测,验证该载体的有效性。
1 材料与方法
1.1 质粒和菌株
基因工程宿主菌E. coli TG1由本实验室保存;质粒pIDM1 (温度敏感、tet抗性) 由德国博士Fuchs TM (Zentralinstitut für Ernährungs-und Lebensmittelforschung (ZIEL) , Abteilung Mikrobiologie,Technische Universität München)馈赠;鸡白痢沙门氏菌CVCC527菌株购自中国兽医药品监察所。
1.2 试剂和工具酶
EcoRⅠ、PstⅠ等限制性内切酶和rTaq DNA聚合酶、dNTPs Mix购自TaKaRa公司;T4 DNA连接酶和 XcmⅠ内切酶购自 New England Biolabs;DNA marker (DL5000、DL2000) 购自东盛公司;AxyPrep质粒DNA小量试剂盒、PCR清洁试剂盒均购自Axygen公司;血液/细胞/基因组DNA提取试剂盒购自北京博大泰克生物有限公司;基因片段引物及接头引物的合成在南京金思瑞生物有限公司完成,测序由华大基因科技股份有限公司完成。
1.3 引物
本实验所用引物名称及序列见表1。
1.4 pIDM-T质粒的构建
1.4.1 XcmⅠ接头引物的设计与合成
参照文献[11]设计一对接头引物 XcmⅠ-up和 XcmⅠ-dn,并在上下游引物的两端分别增加EcoRⅠ位点和PstⅠ位点 (表1),以便克隆入质粒pIDM1的EcoRⅠ位点和PstⅠ位点间,引物由南京金思特有限公司合成。
1.4.2 接头的退火、酶切与纯化
取XcmⅠ-up、XcmⅠ-dn (10 µmol/L) 各10 µL混合,100 ℃下煮沸5 min,缓慢冷却至室温,得到双链接头。对接头进行EcoRⅠ和PstⅠ双酶切并纯化回收。
1.4.3 pIDM1质粒的双酶切
用EcoRⅠ和PstⅠ对pIDM1质粒进行双酶切并回收。
1.4.4 连接
将上述回收的接头酶切产物与 pIDM1/ EcoRⅠ/PstⅠ酶切产物于16 ℃连接,转化E. coli TG1并筛选得到重组质粒pIDM-T。用限制性内切酶 XcmⅠ对重组质粒进行酶切验证和测序分析,测序由华大基因科技股份有限公司完成,测序引物为M13F。1: the EcoR I and the Pst I sites are underlined and the two Xcm I sites are boxed in the Xcm I adapter; 2: the position of the eno and ybdr primers corresponding to the genome sequence of Salmonella gallinarum str. 287/91 (GenBank Accession No. NC_011274.1); 3: the position of universal primers on the constructed pIDM-T vector referred to the Xcm I cassette (for m13F-47up and M13F: upstream of the Xcm I cassette, for m13R-Mdn: downstream of the Xcm I cassette); 4: the theoretically calculated length of DNA fragments amplified together with m13R-Mdn, respectively; 5: the length of DNA amplified from the pIDM-T vector with no insertion.

表1 引物名称及序列Table 1 Names and sequences of primers
1.5 选定基因pIDM-T敲除质粒的构建
1.5.1 目的基因的选取
根据DEG必需基因库 (Database of essential genes) 已有信息和文献报道[8,10,12-13],选取 2个鸡白痢沙门氏菌基因eno和ybdr作为目标敲除基因。eno在DEG库的多种细菌中均为必需基因,ybdr在PEC (Profiling of E. coli Chromosome) 数据库上公布为大肠杆菌的非必需基因,且在本实验室前期鸡白痢沙门氏菌必需基因片段筛选工作中验证为非必需基因。
1.5.2 引物设计与合成
从NCBI上公布的鸡伤寒沙门氏菌基因组序列中,获得选定基因的序列,并应用Primer 5.0设计引物:eno-up和eno-dn,ybdr-up和ybdr-dn,引物序列见表1,引物由南京金思瑞生物有限公司合成。
1.5.3 目的基因片段的扩增与克隆
以S. Pullorum基因组DNA为模板,用上述设计的引物扩增出目的基因片段,将纯化后的基因片段与经纯化的pIDM-T/XcmⅠ载体进行16 ℃连接12~14 h,将连接产物转化大肠杆菌TG1。
1.5.4 阳性克隆筛选
在pIDM-T质粒中具有M13通用引物结合位点,因此选择合成 m13F_47up与 m13R_M-dn引物 (表 1),用于转化克隆的菌落 PCR鉴定。取大肠杆菌转化子稀释液为模板,用上述通用引物进行菌落 PCR检测,筛选阳性克隆 (空载体PCR产物长度为305 bp),进一步抽提阳性克隆质粒,进行酶切鉴定和测序验证。
1.6 pIDM-T质粒复制温敏特性的测定
pIDM-T质粒是在pIDM1基础上插入XcmⅠ位点构建而成的,理论上它与pIDM1质粒一样,在30 ℃培养时正常复制,在37 ℃培养时质粒因不能复制而逐步在子代细胞中丢失,新的细胞将因tet抗性基因丢失而无法在tet平板上存活,只有重组菌因基因组中整合了一拷贝的质粒 (含tet抗性基因) 而能在37 ℃平板上生长。为验证上述推论,以排除因质粒未完全丢失而对IPC值测定的影响,我们开展了如下实验:将pIDM-T空质粒转化鸡白痢沙门氏菌527菌株,挑选阳性转化克隆,30 ℃过夜培养,将过夜培养物稀释后涂布四环素抗性平板,37 ℃培养24~48 h,同时以克隆有非必需 ybdr基因片段的 pIDM-T_ybdr重组质粒转化菌为阳性对照,观察细菌 (重组菌落) 的生长情况。
1.7 IPC值计算及基因的必需性鉴定
将抽提的阳性质粒,电转化至527菌株感受态细胞,涂布含四环素的 LB平板,30 ℃培养24~48 h。挑取单个阳性克隆接种于含四环素的SOC液体培养基中,30 ℃温和振荡培养20~24 h。
为使计数更加精确和方便,对稀释方法进行了摸索。对30 ℃培养条件下的细菌总数计数,尝试了104、105、106、107、108等多个稀释梯度;对37 ℃条件下的重组菌计数,我们进行过2倍、3倍、4倍稀释,比较各个梯度菌液在平板上的生长情况,确定最佳稀释倍数。据前期的摸索用96 孔板将菌液稀释为30倍和120倍,并将稀释菌液50 µL涂布于含四环素的LB平板,每个稀释度做3次重复,将平板置于37 ℃培养24~48 h,计数重组菌的数量。同样再用 96孔板将菌液稀释为105和106倍,将稀释菌液50 µL涂布于含四环素的LB平板,每个稀释度做3次重复,并将平板置于30 ℃培养24~48 h,计数菌液中的细菌总数。
根据公式 (IPC=重组子个数/细菌总个数,即37 ℃平板菌落总数/30 ℃平板菌落总数) 计算出IPC值。据文献报道[8,10],IPC≤1.0×10-6时判定目的基因为必需基因;1.0×10-4≤IPC≤1.0×10-2时判定目的基因为非必需基因。
1.8 鸡白痢沙门氏菌突变株重组插入位点的鉴定
参照鸡伤寒沙门氏菌基因组全序列,分别在目的基因片段的上游200 bp左右设计突变株鉴定引物eno-I-up,ybdr-I-up (表1),将上述鉴定引物与位于pIDM-T载体上的下游引物m13R-Mdn (或上游引物m13F_47up,因T载体克隆片段时可以正反向二种方式插入) 组合,对 37 ℃条件下生长的2个菌落进行PCR鉴定。依据能否扩增出特异的基因片段及片段的测序结果鉴定突变株IDM插入位点的正确性。
2 结果
2.1 pIDM-T质粒的构建和鉴定
将XcmⅠ接头插入pIDM1的EcoRⅠ和PstⅠ后获得pIDM-T重组质粒,为验证接头已正确插入,用XcmⅠ酶切重组质粒,获得1条约3.9 kb的片段;而未插入接头的 pIDM1原始质粒不能被XcmⅠ酶所消化 (图1)。用引物M13-F对已构建的pIDM-T质粒进行测序,结果与预期一致。

图1 pIDM-T重组质粒的酶切鉴定Fig. 1 Gel electrophoresis of the recombinant vector pIDM-T identified by enzyme digestion. M: marker (DL5000); 1: pIDM-T digested with XcmⅠ; 2: pIDM1 digested with XcmⅠ; 3: pIDM-T plasmid.
2.2 选定基因pIDM-T敲除质粒的构建
eno和 ybdr基因扩增片段的预期大小分别为:429 bp和436 bp,电泳结果显示与预期相符(图2)。用通用引物m13F_47up与m13R_Mdn对pIDM-T载体克隆的转化子进行菌落PCR检测,理论上阳性克隆片段条带大小为目的基因克隆片段加 300 bp (空载体扩增出的条带大小为305 bp),对应地eno、ybdr阳性克隆的扩增长度分别约为730 bp和740 bp,电泳检测结果与预期一致 (图略)。提取初步鉴定为阳性克隆的质粒,用EcoRⅠ和PstⅠ双酶切再次验证,电泳结果可见两条带,小片段与插入片段大小一致 (图3)。
2.3 pIDM-T质粒复制温敏特性的测定
据文献报道[14],pIDM1在沙门氏菌细胞中的质粒拷贝数约为300个,尽管细菌在37 ℃培养时 pIDM1质粒不复制,但它可以继续随细胞分裂而分配,因此,pIDM1质粒可最多分配到200多个子代细胞中。如果细菌基因组上没有重组的质粒拷贝,新的细菌细胞中将因质粒丢失而无法在四环素平板上存活。pIDM-T质粒是在pIDM1质粒的基础上插入 XcmⅠ酶切位点构建而成的,理论上它与 pIDM1质粒的温敏特性是一样的,因此,如果pIDM-T或其衍生质粒不与基因组发生有效的重组,tet基因传递给子代细胞的数量是有限的。

图2 目的片段扩增Fig. 2 The amplification of the target genes. M: marker (DL2000); 1: amplification without template; 2: eno fragment; 3: ybdr fragment.

图3 阳性质粒的双酶切鉴定Fig. 3 The double digestion of the target plasmid. M: marker (DL5000); 1: EcoRⅠ/PstⅠdouble digestion of pIDM-T_eno; 2: EcoRⅠ/PstⅠdouble digestion of pIDM-T_ybdr.
为验证上述推论,在实验时我们将pIDM-T空质粒转化鸡白痢沙门氏菌527菌株,阳性转化菌 30 ℃过夜培养后稀释涂布于四环素抗性平板,37 ℃培养约36 h后,观察细菌 (重组菌落)的生长情况,发现在pIDM-T空质粒转化菌涂布的平板上仅见到较淡的菌苔背景,无单个的重组菌落 (图4A);而平行开展的pIDM-T_ybdr重组质粒转化菌则在较淡的菌苔背景上见到明显的重组菌落 (图4B,黑色箭头标示)。
由此可见,pIDM-T质粒在37 ℃培养时会逐步丢失,不会影响IPC值的测定。
2.4 IPC值计算及选定基因的必需性鉴定
将2个阳性重组质粒分别电转化527菌株,将转化菌株接种LB液体30 ℃增菌,菌液稀释涂板后分别置于30 ℃和37 ℃培养。30 ℃培养用于计数菌落总数,而37 ℃培养用于计数重组子数。根据公式IPC=重组子数/细菌总数=37 ℃平板菌落总数/30 ℃平板菌落总数,计算得eno和ybdr的IPC值分别为1.92×10-7和1.64×10-4(表2)。从测定数值看出,eno基因片段的重组率低于1.0×10-6,鉴定为必需基因,而ybdr基因片段重组率较高,鉴定为非必需基因,与预期结果一致。

图4 pIDM-T及其衍生质粒转化菌的37 ℃平板培养结果Fig. 4 Growth of the transformants of pIDM-T and its derivative plasmid in 37 ℃. (A) The growth of the 527 strain with pIDM-T plasmid on tet plate after incubated in 37 ℃ for 36 hours. (B) The growth of the 527 strain with pIDM-T_ybdr plasmid on tet plate after incubated in 37 ℃ for 36 hours. Black arrows point to recombinant colonies grown on the plate.
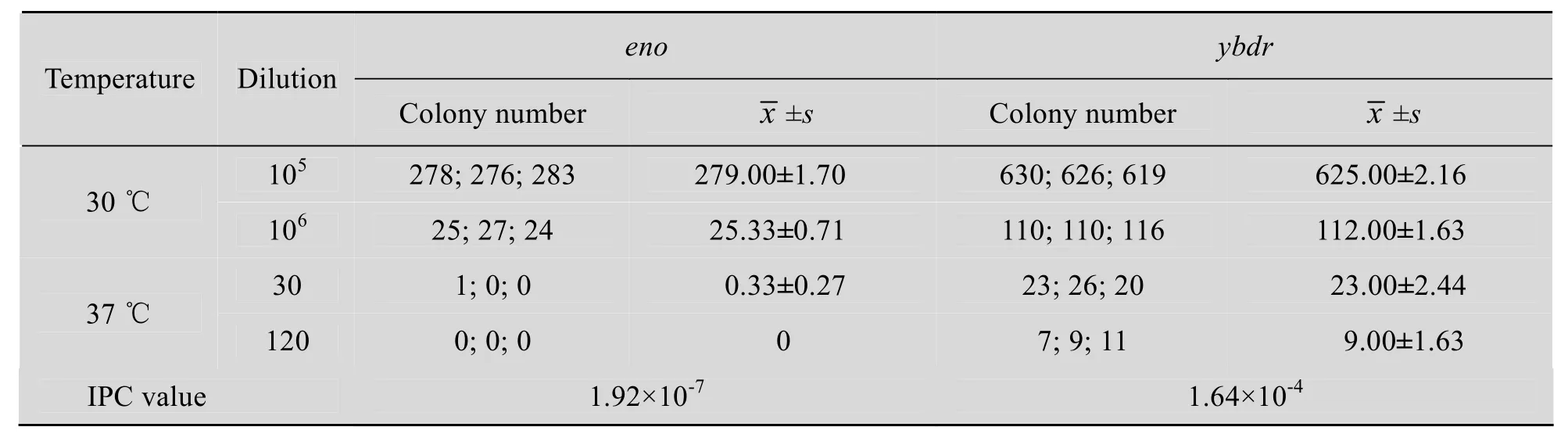
表2 菌落计数及IPC值测定Table 2 The colony number and IPC value
2.5 鸡白痢沙门氏菌突变菌株重组插入位点的鉴定
携带非必需的 ybdr基因片段的pIDM-T_ybdr质粒电转化至527菌株后,目的基因片段可与细菌基因组 DNA同源片段发生重组,pIDM-T质粒骨架插入到对应基因中,使目的基因被敲除,同时使细菌基因组中携带1拷贝的tet抗性基因 (原理图见图5)。尽管37 ℃条件下pIDM-T_ybdr质粒不能复制,逐渐丢失,由质粒表达的 tet抗性也随之消失,但发生了同源重组的细菌基因组上仍含有1拷贝的tet基因,因此重组菌可以在四环素平板上存活。然而,携带eno基因片段的pIDM-T_eno质粒电转化至 527菌株后,因eno基因的必需性,发生特异性同源重组的菌株因eno基因的敲除而死亡,因此理论上不可能出现eno基因中插入质粒骨架和tet基因的菌株。
为验证上述分析的正确性,我们从37 ℃培养的pIDM-T_ybdr质粒转化平板中,随机挑选了2个菌落,用 ybdr-I-up上游引物分别与m13R-Mdn和m13F-47up引物组合进行PCR扩增,结果用ybdr-I-up与m13R-Mdn引物组合时扩增出约750 bp的片段,ybdr-I-up与m13F-47up引物组合扩增无产物 (图 6)。对扩增出的约750 bp的片段进行测序,结果显示该片段含有一段ybdr基因序列和小段载体序列 (144 bp),与预期一致,说明构建的SalΔybdr 527突变株是由于pIDM-T载体克隆的ybdr基因片段经IDM方式特异性地同源重组的结果。检索GenBank中的基因功能注释,ybdr基因编码一个脱氢酶。可能由于鸡白痢沙门氏菌527菌株具有多种该脱氢酶的同工酶的缘故,在 LB液体和 LB平板培养SalΔybdr 527突变株和原始野生株时,未发现二者具有明显的生长特性差异。突变株的其他表型鉴定有待进一步开展。

图5 SalΔybdr突变株构建原理图Fig. 5 Construction of the SalΔybdr mutant strain by homologous recombination. A 436 bp fragment of ybdr gene was cloned into pIDM-T, and the resulting IDM mutant SalΔybdr carries a truncated gene followed by linearized plasmid pIDM-T and rest part of the gene.
对eno基因敲除平板筛选到的1个tet抗性菌落,我们用引物eno-I-up分别与m13R-Mdn和m13F-47up引物组合对其进行 PCR扩增鉴定,结果无任何条带 (图6),对野生菌株菌液的对照PCR扩增也无任何产物,因此可初步确定质粒没有特异性地插入eno基因中。进一步以该菌培养物的稀释液为模板,以eno-up和 eno-dn为引物扩增eno基因片段,电泳检测获得与预期一致的DNA条带 (约430 bp,图略),测序结果进一步显示eno基因为野生型,说明eno基因没有被插入突变。将该菌抽提质粒,电泳结果未检测到与pIDM-T质粒大小相近的小质粒 (约3.9 kb,图略)。由此推测,该克隆可能是质粒随机重组至细菌基因组的结果,具体重组位置有待进一步测定。
3 讨论
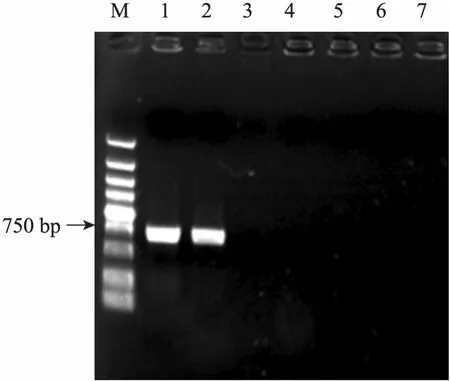
图6 突变菌株的PCR鉴定Fig. 6 PCR verification of the mutant strains. M: marker (DL5000); 1,2: SalΔybdr mutant strain amplified by ybdr-I-up and m13R-Mdn; 3: SalΔeno mutant strain amplified by eno-I-up and m13R-Mdn; 4: SalΔeno mutant amplification by eno-I-up and m13F-47up; 5: SalΔybdr mutant amplification by ybdr-I-up and m13F-47up; 6,7: wild type control.
Taq DNA聚合酶具有末端转移酶活性,在扩增DNA的3′末端非模板依赖地加上一个腺苷酸。为解决PCR产物的克隆,需要在线性化载体的3′末端多加一个的脱氧胸腺嘧啶核苷 (T),这就是TA克隆,相应的载体通常被称为 T-载体[15-16]。商品 T载体多数是在平末端上加 T而成,而XcmⅠ识别CCA (N5/N4) TGG序列,切割后产生单个核苷酸突出的3¢末端,且切割位点附近的碱基可以简并[16]。将识别序列的第8位设为T,重组质粒在两个反向XcmⅠ序列接头处经XcmⅠ切割,便可获得线性化的T载体。
Knuth等[8]用pIDM1质粒构建了鼠伤寒沙门氏菌的基因文库,通过 IDM的方法鉴定了鼠伤寒沙门氏菌ATCC14028的必需基因,我们采用类似的方法构建了鸡白痢沙门氏菌 527菌株文库,并筛选了 527菌株的必需基因 (结果待发表)。pIDM1可用于克隆随机片段,构建文库,并有效地应用于必需基因的大规模筛选和鉴定,但在实际应用时该质粒需首先经 KpnⅠ酶切线性化,然后用热敏磷酸酶 (Antarctic phosphates)脱磷处理,防止自身环化[17],操作繁琐,成功率低;另一方面它不能直接克隆 PCR产物,因此PCR产物克隆前同样需要酶切。为方便地克隆PCR产物,提高效率,以用于特定基因的敲除,我们在 pIDM1载体的多克隆位点插入了两个串联XcmⅠ接头,对pIDM1质粒进行了T载体改造,经检测正确。构建的pIDM-T质粒经XcmⅠ酶切后即可成为T载体,能直接快速有效地克隆未经处理的单个目标PCR产物。由于T载体采用内切酶法制备,保证了质量的稳定,且成本低廉,适合普通实验室使用。
理论上,pIDM-T与 pIDM1质粒一样,在37 ℃培养时不复制,存在于细菌中的原始质粒拷贝将随细菌的分裂而分配给有限的子代细胞,若质粒不与基因组发生有效的重组,经一定代数后,pIDM-T质粒将从新增殖的细胞中丢失而使细菌无法在四环素平板上存活。为验证上述推论,我们将pIDM-T空质粒和pIDM-T_ybdr重组质粒同时转化鸡白痢沙门氏菌527菌株,通过观察两种质粒转化菌在 37 ℃ tet平板上的生长情况,我们发现:pIDM-T空质粒的转化菌在平板上仅形成淡淡的菌苔背景,未见单个重组菌落;而 pIDM-T_ybdr重组质粒转化菌则在较淡的菌苔背景上生长有明显的重组菌落。由此可见,pIDM-T质粒在37 ℃培养时会逐渐丢失,转化菌在四环素平板上才出现了有限增殖,因此,它不影响IPC值的测定。
为使细菌总数和重组菌计数更加精确和方便,以获得准确的IPC值,我们对稀释方法进行了摸索。对30 ℃培养条件下的细菌总数计数,我们曾尝试 104、105、106、107、108等多个 10倍梯度稀释,结果发现在设定的 96孔板培养条件下50 µL菌液中的细菌总数在2.5×106~2.1×108之间,105和 106倍两个稀释梯度较为合适;对37 ℃条件下的重组菌计数,我们曾尝试10、20、40、80和160倍 (经10倍稀释后再倍比稀释),10、30、90和270倍 (10倍稀释后再3倍稀释) 及30、120和480倍 (30倍稀释后再4倍稀释) 等多个稀释梯度,结果发现稀释倍数偏低会因为形成菌苔而掩盖了重组菌落的识别,稀释过高则平板上无菌落生长,而30和120倍二个梯度较为适合。
与pIDM1质粒一样,pIDM-T可应用于特定基因的敲除及必需性的鉴定。为验证pIDM-T的有效性,我们选取eno和ybdr两个基因,运用插入复制突变法在鸡白痢沙门氏菌527菌株中进行了必需性鉴定和敲除。将eno和ybdr扩增片段克隆于线性化的pIDM-T载体,转化大肠杆菌TG1,经鉴定获得带有目的片段的重组质粒。将携带有目的片段的重组质粒电转化至527菌株,将转化子增菌后稀释涂板,并分别置于30 ℃和37 ℃培养,计数重组子和菌落总数,根据公式计算得出eno的IPC值为1.92×10-7,鉴定为必需基因;ybdr的IPC值为1.64×10-4,鉴定为非必需基因。实验结果与 DEG必需基因库 (Database of essential genes)、大肠杆菌 PEC(Profiling of E. coli Chromosome)数据库的已有信息,以及文献报道[12-13]和本实验室前期实验结果相符。
为确认IDM敲除非必需的ybdr基因的插入位点的正确性,以及IDM无法敲除必需基因eno,我们从ybdr基因敲除的筛选平板上随机挑取了2个克隆,PCR扩增质粒插入ybdr基因的交界处片段,并经测序证实pIDM-T质粒骨架已插入在ybdr基因所在的细菌基因组特定位点,由此说明,ybdr基因已通过IDM重组被特异性地敲除。ybdr基因是一个脱氢酶编码基因[18-19],可能由于鸡白痢沙门氏菌527菌株具有多种该脱氢酶的同工酶的缘故,我们未发现SalΔybdr 527突变株和原始野生株在LB液体和LB平板生长特性的差异。
我们对eno基因敲除筛选平板上生长的唯一1个抗性菌落同样进行了PCR扩增,结果无任何条带;进一步对该菌落的eno基因片段进行扩增,扩增产物的电泳检测和测序结果均显示eno基因为野生型,由此说明eno基因未被pIDM-T质粒骨架插入而破坏。对该抗性菌培养并抽提质粒,结果也未检测到与 pIDM-T质粒大小相近的质粒,因此我们推测该菌落可能是质粒随机重组至细菌基因组的结果。据报道,Knuth等[8]应用pIDM1质粒鉴定鼠伤寒沙门氏菌 257个必需基因时,超过1/3的克隆有非特异性重组现象 (即IPC值大于零),我们在开展鸡白痢沙门氏菌必需基因克隆鉴定时也有类似的现象 (结果待发表),可见这种随机重组现象是较普遍的,它可能受基因片段长度等多种因素影响[20],但因IPC值的数量级不同,它不影响基因必需性的鉴定。
由此可见,本实验构建的pIDM-T质粒是一个有效快速的基因敲除载体,它可以快速克隆PCR产物,并应用IDM方法快速有效地鉴定特定基因的必需性,构建非必需基因的突变株,为鸡白痢沙门氏菌基因功能研究提供了一种有效的手段。
REFERENCES
[1] Tenno T, Goda N, Tateishi Y, et al. Highthroughput construction method for expression vector of peptides for NMR study suited for isotopic labeling. Protein Eng Des Sel, 2004, 17(4): 305−314.
[2] Li JH, Han CQ, Deng J, et al. Construction and functional analysis of a common gene targeting vector with double-selection markers. Chin J Biotech, 2010, 26(12): 1696−1703.李军华, 韩翠芹, 邓捷, 等. 双筛选标记打靶载体的构建及其功能鉴定. 生物工程学报, 2010, 26(12): 1696−1703.
[3] Jiang N, Wang YC, Ma ZH, et al. A novel temperature sensitive plasmid-based method for deletion of chromosomal genes. China Biotechol, 2010, 30(3): 85−89.姜娜, 王艳春, 马志宏, 等. 一种基于温敏质粒的新型基因敲除方法. 中国生物工程杂志, 2010, 30(3): 85−89.
[4] Blaby IK, Phillips G, Blaby-Haas CE, et al. Towards a systems approach in the genetic analysis of Achaea: accelerating mutant construction and phenotypic analysis in Haloferax volcanii. [EB/OL]. [2012-2-25]. http://www.hindawi.com/journals/ arch/2010/426239/.
[5] Baek SH, Rajashekara G, Splitter GA, et al. Denitrification genes regulate Brucella virulence in mice. J Bacteriol, 2004, 186(18): 6025−6031.
[6] Qiao F, Chen ZL, Wang YF, et al. Construction of plasmid pUC19K and its application in Brucella mutant construction. China Biotechnol, 2007, 27(12): 1−5.乔凤, 陈泽良, 王玉飞, 等. pUC19K质粒的构建及其在布鲁氏菌突变株构建中的应用. 中国生物工程杂志, 2007, 27(12): 1−5.
[7] Hu Y, Cong YG, Qiu RR, et al. Construction of a bcfD gene knock-out mutant of Salmonella enterica serovar typhi. Biotechnology, 2011, 21(3): 10−13.胡勇, 丛延广, 邱荣蓉, 等. 伤寒沙门菌 bcfD基因敲除突变株的构建. 生物技术, 2011, 21(3): 10−13.
[8] Knuth K, Niesalla H, Hueck C J, et al. Large-scale identification of essential Salmonella genes by trapping lethal insertions. Mol Microbiol, 2004, 51(6): 1729−1744.
[9] Biswas I, Vanger V, Eerlich SD. Efficiency of homologous intermolecular recombination at different locations on the Bacillus subtilis chromosome. J Bacteriol, 1992, 174(17): 5593−5596.
[10] Klumpp J, Fuchs TM. Identification of novel genes in genomic islands that contribute to Salmonella typhimurium replication in macrophages. Microbiology, 2007, 153(4): 1207−1220.
[11] Park HK, Zeng CY. Construction of an Xcm I-generated T vector bearing green fluorescent protein marker for direct cloning of PCR products. Anal Biochem, 2007, 360(1): 144−145.
[12] Cohen R, Yokoi T, Holland JP, et al. Transcription of the constitutively expressed yeast enolase gene ENO1 is mediated by positive and negative cis-acting regulatory sequences. Mol Cell Biol, 1987, 7(8): 2753−2761.
[13] Burnett ME, Liu J, Conway T. Molecular characterization of the Zymomonas mobilis enolase (eno) gene. J Bacteriol, 1992, 174(20): 6548−6553.
[14] Fuchs TM, Klumpp J, Przybilla K. Insertionduplication mutagenesis of Salmonella enterica and related species using a novel thermosensitive vector. Plasmid, 2006, 55(1): 39−49.
[15] Zhou MY, Clark SE, Gomez-Sanchez CE. Universal cloning method by TA strategy. Biotechniques, 1995, 19(1): 34−35.
[16] Jo C, Jo SA. A simple method to construct T-vectors using Xcm I cassettes amplified by nonspecific PCR. Plasmid, 2001, 45(1): 37−40.
[17] Sun CL, Li YW, Wang Y, et al. Construction of a pUC19-T Vector based on Xcm I. J Anhui Agric Sci, 2011, 39(17): 10182−10184.孙程龙, 李业伟, 王颖, 等. 基于 Xcm I酶切的pUC19-T载体的构建. 安徽农业科学, 2011, 39(17): 10182−10184.
[18] Riley M, Abe T, Arnaud MB, et al. Escherichia coli K-12: a cooperatively developed annotation snapshot-2005. Nucleic Acids Res, 2006, 34(1): 1−9.
[19] Erickson KD, Detweiler CS. The Rcs phosphorelay system is specific to enteric pathogens/commensals and activates ydeI, a gene important for persistent Salmonella infection of mice. Mol Microbiol, 2006, 62(3): 883−894.
[20] Zhou JG, Hong X, Huang CF. Recombineering and its application. Acta Gen Sin, 2003, 30(10): 983−988.周建光, 洪鑫, 黄翠芬. 重组工程及其应用. 遗传学报, 2003, 30(10): 983−988.
猜你喜欢
复旦学报(医学版)(2021年4期)2021-08-05
数学大王·中高年级(2021年4期)2021-04-27
装备制造技术(2020年3期)2020-12-25
家庭影院技术(2019年8期)2019-08-27
食品与机械(2019年1期)2019-03-30
兽医导刊(2016年12期)2016-05-17
现代食品(2016年24期)2016-04-28
食品工程(2015年3期)2015-12-07
电测与仪表(2015年20期)2015-04-09
特产研究(2014年4期)2014-04-10
