
肺炎支原体P1蛋白羧基端基因片段的克隆及原核表达
2012-01-27 06:57王云龙刘小聪李玉林王国强孙新城董彩文王继创邓黎黎李恒思
动物医学进展 2012年6期
王云龙,刘小聪,李玉林,王国强,孙新城,董彩文,程 蕾,王继创,邓黎黎,李恒思
(1.新乡医学院,河南新乡450003;2.郑州职业技术学院,河南郑州450121;3.河南省生物工程技术研究中心,河南郑州450001)
肺炎支原体P1蛋白羧基端基因片段的克隆及原核表达
王云龙1,2,刘小聪1,李玉林3,王国强3,孙新城3,董彩文3,程 蕾3,王继创3,邓黎黎3,李恒思3
(1.新乡医学院,河南新乡450003;2.郑州职业技术学院,河南郑州450121;3.河南省生物工程技术研究中心,河南郑州450001)
根据GenBank公布的肺炎支原体(Mycoplasmapneumoniae,Mp)P1蛋白羧基端序列(AF290001.1),设计合成2对特异性引物,采取套式PCR方法,以肺炎支原体培养物中提取的DNA为模板,扩增肺炎支原体P1蛋白羧基端基因序列,将其克隆至p ET32a表达载体,构建重组质粒p ET32a/P1(3 802~4 695),转化大肠埃希菌BL21-gold,测序验证后IPTG诱导表达,经SDS-PAGE表明重组蛋白表观分子质量与预期一致,可溶性表达产物经Western blot证实重组蛋白具有免疫反应性。
肺炎支原体;P1蛋白;套式PCR;原核表达
肺炎支原体(Mycoplasmapneumoniae,Mp)是引起人类原发性肺炎的常见病原体之一。Mp感染广泛存在,寒冷季节发病率较高,每4年~5年可引起一次地区性暴发流行[1]。Mp感染以儿童多见,据文献报道我国Mp感染率在流行高峰期可达33%~38%[2-4]。除呼吸道急性感染症状外,亦可诱发哮喘、阻塞性肺疾病的急性发作及导致心血管、血液、中枢神经等多系统损害[5-6],临床表现缺乏特异性,不利于诊断。因此,建立早期、快速的诊断方法对指导临床治疗有重要意义。
Mp感染的免疫学发病机制研究显示,位于黏附细胞器顶端表面的P1黏附蛋白是其主要功能结构,含有多个抗原决定簇,可激发机体的免疫反应[7]。但作为制备 Mp诊断试剂盒的候选蛋白,P1黏附蛋白结构与生殖道支原体、真核生物结构蛋白结构存在一定相似性,以完整的Pl黏附蛋白制备诊断试剂时,可能出现交叉反应。国外学者对其进行拓扑结构分析发现,在P1黏附蛋白结构中与黏附有关的区域及特异的Mp P1黏附蛋白抗原决定簇主要位于羧基端[8]。因此,可选择该亚单位用于制备Mp诊断试剂。
本试验根据GenBank中肺炎支原体的基因序列设计引物,采用套式PCR方法扩增P1蛋白羧基端序列,并对其进行序列分析及原核表达。表达产物经纯化和生物活性鉴定后,可用于检测方法的建立,并为基因工程疫苗的研究提供新的途径。
1 材料与方法
1.1 材料
1.1.1 肺炎支原体培养物、菌株与载体 克隆菌Top10F'、表达载体p ET32a及表达菌BL21-gold为河南省生物工程技术研究中心提供。
1.1.2 主要试剂 DNA提取试剂盒为Axygen公司产品;Taq酶、D2000 DNA Marker、蛋白分子质量标准为天根生化科技(北京)有限公司产品;质粒提取试剂盒、限制性内切酶BamHI、XhoI及T4连接酶为宝生物工程(大连)有限公司产品;琼脂糖为Biowest公司原装;其他常规试剂均为国产分析纯产品。
1.1.3 肺炎支原体阳性血清 为郑州大学第一附属医院提供。
1.2 方法
1.2.1 引物的设计与合成 根据GenBank公布的人肺炎支原体P1蛋白羧基端基因序列(AF290001.1),参考黄劲松[9]和赵芝娜[10]的肺炎支原体P1蛋白羧基端基因特异引物,对引物进行重新设计,合成2对特异性引物P1/P2和P3/P4。

其中,P1/P2为外引物,P3/P4为内引物,采用套式PCR方法,最终扩增片段为894 bp。引物由上海生工生物工程技术服务有限公司合成。
1.2.2 肺炎支原体基因组的提取 按照试剂盒说明书操作。
1.2.3 P1黏附蛋白羧基端序列的扩增 第1次PCR扩增反应体系的总体积为30μL:TaqDNA聚合酶 0.4μL,20 倍缓冲液 1.5μL,Mg2+(50 mmol/L)0.6μL,d NTP(10 mmol/L)0.5μL,P1/P2引物 (10 pmol/L )各 0.5μL,模板 (基因组DNA)2μL,去离子水补足30μL。反应条件为:94℃5 min;94℃1 min,58℃1 min,72℃1.5 min,35个循环;72℃5 min。第2次PCR扩增反应体系总体积为30μL:TaqDNA聚合酶0.4μL,20倍缓冲液1.5μL,Mg2+(50 mmol/L)0.6μL,d NTP(10 mmol/L)0.5μL,P3/P4引物(10 pmol/L)各0.5 μL,第一次扩增产物1μL,去离子水补足30μL。反应条件为:94℃5 min;94℃1 min,60℃1 min,72℃1.5 min,35个循环;72℃5 min。反应结束后于10 g/L琼脂糖凝胶上电泳,分析扩增产物。
1.2.4 原核表达载体的构建 用DNA回收试剂盒对PCR产物进行回收,将回收的片段和载体p ET32a同时进行BamH I、XhoI双酶切,连接后转化E.coilTop10F'感受态细胞。重组菌经培养后提质粒,质粒经PCR及BamH I、XhoI双酶切鉴定为阳性者转化BL21-gold感受态细胞,挑选单克隆扩大培养并送上海生工生物工程技术服务有限公司测序,以确定目的片段序列及插入位置的正确性。测序结果与GenBank公布的序列进行对比,若鉴定正确,命名重组质粒为p ET32a/P1(3802~4695)。
1.2.5 重组蛋白的诱导表达、纯化及 SDS-PAGE分析 取鉴定正确的菌液活化过夜,平行接种至2支3.5 m L的LB培养基(含氨苄西林)中,37℃培养至OD600 nm为0.6时,取其中一支用终浓度为0.1 mmol/L的IPTG 进行诱导,另一支做对照。4 h后每支各取1 m L菌液制蛋白样,进行SDSPAGE。
样品处理方法:取1 m L菌液于离心管中,12 000 r/min离心2 min,弃上清,100μL PB(p H 7.6,0.05 mol/L)悬菌,超声波破碎后12 000 r/min离心2 min,分离上清和沉淀。取30μL上清加入等体积2×SDS上样缓冲液,沉淀中加30μL PB(p H7.6,0.05 mol/L)混匀后加入等体积2×SDS上样缓冲液,沸水浴5 min,分别制成上清和沉淀的蛋白样品。取上述蛋白样品进行SDS-PAGE,鉴定表达产物的分子质量,分析蛋白可溶性。
1.2.6 重组蛋白的 Western blot鉴定 取上述重组融合蛋白样品进行SDS-PAGE,同时设空质粒对照。以恒定电流将蛋白质转移到硝酸纤维素膜上。硝酸纤维素膜用PBST清洗5次后以10 m L/L BSA封闭2 h;加入人肺炎支原体抗体阳性血清,37℃温育1.5 h,PBST洗膜;加入辣根过氧化物酶标记的鼠抗人IgG,37℃温育2 h,清洗后加入DAB显色液显色,待出现条带后立即用PBST终止显色,观察结果并拍照。
2 结果
2.1 P1蛋白羧基端基因的扩增
电泳结果显示,第一次扩增得到了一条1 044 bp的条带,第二次扩增得到了一条约894 bp的条带,与预期结果相符(图1)。
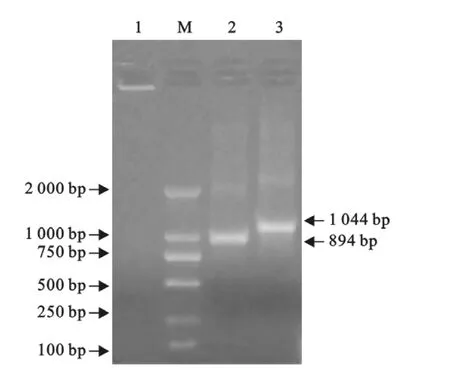
图1 Mp P1蛋白羧基端基因的PCR扩增结果Fig.1 PCR identification of C-terminal gene fragment of Mp P1
2.2 p ET32a/P1(3 802~4 695)重组质粒的双酶切鉴定
重组质粒PET32a/P1(3 802~4 695)经BamHⅠ和XhoⅠ双酶切得到约894 bp的条带,与预期结果相符(图2)。
2.3 P1蛋白羧基端基因的测序鉴定
测序结果与已公布P1黏附蛋白羧基端核苷酸序列(AF290001.1)一致性为99.54%,氨基酸序列一致性为100%。
2.4 重组蛋白SDS-PAGE分析
重组菌在37℃条件下,经终浓度为0.1 mmol/L的IPTG诱导表达4 h后,超声破碎提取蛋,白大小约为50 ku,与预期结果相符。电泳结果显示重组蛋白呈可溶性表达(图3)。
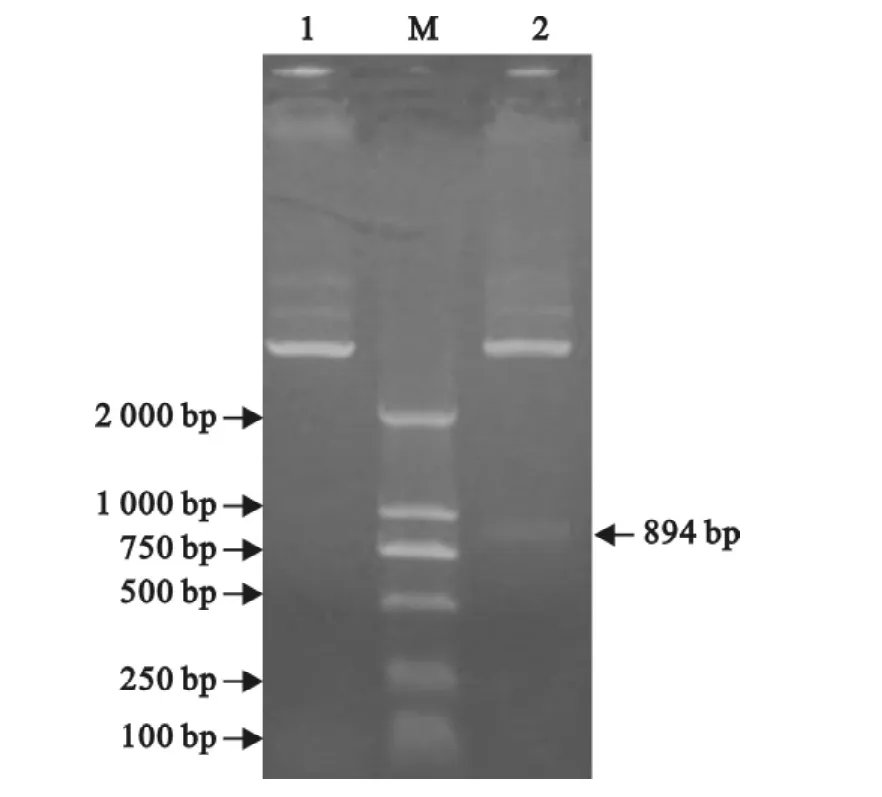
图2 重组质粒p ET32a/P1(3 802~4 695)双酶切鉴定Fig.2 Identification of p ET32a/P1(3 802-4 695)by restriction enzyme digestion
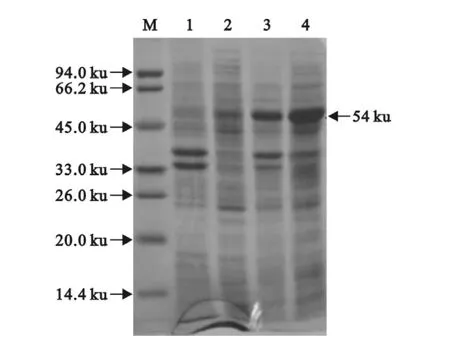
图3 重组菌诱导表达产物的SDS-PAGE分析Fig.3 SDS-PAGE analysis of expression products of BL21-gold/pET32a/P1
2.5 重组蛋白的免疫反应性检测
用肺炎支原体抗体阳性血清检测重组蛋白的免疫原性,显色后在硝酸纤维膜上出现单一条带(图4)。
2.6 表达产物纯化
目的蛋白分子质量约为32 ku,带有 His-tag、STag和Trx-tag标签的融合蛋白分子为可溶性蛋白,蛋白分子质量为50 ku左右,与预期相符,Ni2+亲合层析纯化后进行SDS-PAGE分析,凝胶扫描显示纯度达90%以上(图5)。

图4 表达产物的免疫印迹分析Fig.4 Analysis of expressed products by Western blot

图5 SDS-PAGE分析表达产物的纯化结果Fig.5 SDS-PAGE analysis of purifid expressed products
3 讨论
肺炎支原体是原发性肺炎的病原体。感染后以持续性剧烈干咳为主要临床表现,症状不典型,很难与细菌、病毒所致呼吸道感染相区别。除呼吸系统症状往外,Mp感染尚能诱发肺外并发症,如心绞痛、肾炎、肺炎支原体脑炎、溶血性贫血、皮疹、关节炎等[11],症状复杂,不利于诊断。为避免延误治疗,建立快速、早期、准确的诊断方法尤为重要[12]。
免疫学研究证实黏附是Mp致病的关键步骤,已有的研究表明P1蛋白羧基端是肺炎支原体的主要黏附区,含有较多的P1蛋白抗原决定簇[13]。黄劲松等[9]对不同亚型的 Mp进行分析,发现这段基因片段在不同亚型的Mp中同源性100%。因此P1蛋白羧基端是制备基因工程疫苗和快速诊断试剂盒的理想材料。
为提高PCR的灵敏度和特异性,本试验采用套式PCR方法,不仅可提高扩增倍数从而提高PCR敏感性,还可通过二次扩增降低非特异性反应。测序结果表明该片段长度为894 bp,编码298个氨基酸。Western blot表明表达产物具有良好的免疫反应性,为下一步研发疫苗及制备快速检测试剂奠定了基础。
[1] Blasi F,Tarsia P,Alibcrti S,et al.ChlamydiapneumoniaeandMycoplasmapneumoniae[J].Semin Respir Crit Care Med,2005,26(6):617-624.
[2] 辛德莉,李 贵,李 娟,等.北京地区肺炎 MP肺炎的流行状况[J].实用儿科临床杂志,2006,21(16):1042-1055.
[3] 王雪峰,董 丹,刘 芳,等.小儿肺炎840例常见病原分析[J].中国实用儿科杂志,2005,20(4):239-241.
[4] 汤文红,曹秀章.小儿呼吸道肺炎支原体感染发病趋势及临床分析[J].临床儿科杂志,2005,23(8):562-563.
[5] Macdowell A L,Bacharier L B.Infectious triggers of asthma[J].Immunol Allerg Clin North Am,2005,25(1):45-66.
[6] Wisniewska-Ligier M,Wozniakowska-Gesicka T,Sobanska A,et al.Extrapulmonary complications ofMycoplasmapneumoniaeinfections[J].Przegl Lek,2003,60(12):832-835.
[7] Razin S.Adherence of pathogenic mycoplasmas to host cells[J].Biosci Rep,1999,19:367-372.
[8] Gerstenecker B,Jacobs E.Topological mapping of the P1-adhesin ofMycoplasmapneumoniaewith adherence-inhibiting monoclonal antibodyies[J].J Gen Microbiol,1990,136(3):471.
[9] 黄劲松,黄捷勤,唐启慧,等.肺炎支原体P1蛋白羧基端基因片段在大肠杆菌中的表达及应用研究[J].中国生物工程杂志,2010,30(11):65-69.
[10] 赵芝娜,王桂珍,董占双,等.肺炎支原体P1黏附蛋白基因的克隆与表达[J].中国人兽共患病杂志,2005,21(6):470-472.
[11] 赵顺英.肺炎支原体感染的临床表现和肺外并发症[J].实用临床儿科杂志,2007,22(4):249-250
[12] Chang Y T,Yang Y H,Chiang B L,et al.The significance of a rapid cold hemagglutination test for detectingMycoplasmainfections in children with asthma exacerbation[J].J Microbiol Immunol Infect,2006,39(1):28-32.
[13] Krause D C.Mycoplasmapneumoniaecytadherence:unravelling the tie that binds[J].Micobiology,1996,20:247-253.
Cloning and Prokaryotic Expression of C-terminal Gene Fragment of P1 Protein ofMycoplasmapneumonia
WANG Yun-long1,2,LIU Xiao-cong1,LI Yu-lin3,WANG Guo-qiang3,SUN Xin-cheng3,DONG Cai-wen3,CHENG Lei3,WANG Ji-chuang3,DENG Li-li3,LI Heng-si3
(1.XinxiangMedicalUniversity,Xinxiang,Henan,450003,China;2.ZhengzhouTechnicalCollege,Zhengzhou,Henan,450121,China;3.HenanBiotechnologyResearchCenter,Zhengzhou,Henan,450001,China)
According to the published nucletide sequence of C-terminal gene fragment of P1 protein ofMycoplasmapneumoniain GenBank(AF290001.1),2 pairs of specific primers were designed and the total DNA was extracted from Mp.The fragment amplified by nested PCR was cloned into prokaryotic expression vector pET-32a and transformed into BL21-gold.After the recombinant plasmid pET32a/P1(3 802-4 695)was sequenced and compared with other strains in GenBank,it was induced by IPTG.The recombinant protein was identified by SDS-PAGE and Western blot.SDS-PAGE analysis revealed that the expressed protein was identical to C-terminal gene fragment of P1 protein in molecular weight,Western-blot showed that the expressed protein can be recognized byMycoplasmapneumoniapositive serum.
Mycoplasmapneumonia;P1 protein;nested PCR;prokaryotic expression
S852.62
A
1007-5038(2012)06-0045-04
2012-02-16
王云龙(1962-),男,河南孟津人,教授,主要从事细胞生物学研究。
猜你喜欢
江西农业学报(2021年4期)2021-04-20
传染病信息(2021年6期)2021-02-12
科海故事博览·下旬刊(2019年6期)2019-04-16
兽医导刊(2019年1期)2019-02-21
兽医导刊(2019年1期)2019-02-21
中国生殖健康(2018年4期)2018-11-06
河南畜牧兽医(2017年12期)2017-11-13
妈妈宝宝(2017年3期)2017-02-21
西南医科大学学报(2015年1期)2015-08-22
中国当代医药(2015年9期)2015-03-01
