
GPU引发的计算化学革命
2011-12-11 09:08鲍建樟丰鑫田于建国
物理化学学报 2011年9期
鲍建樟 丰鑫田 于建国,*
(1北京师范大学管理学院系统科学系,北京100875;2北京师范大学化学学院,北京100875)
GPU引发的计算化学革命
鲍建樟1丰鑫田2于建国2,*
(1北京师范大学管理学院系统科学系,北京100875;2北京师范大学化学学院,北京100875)
综述了图形处理器(GPU)在计算化学中的应用和进展.首先简单介绍了GPU在科学计算中应用的发展,然后分别详细讲述了迄今几个使用GPU和CUDA(compute unified device architecture,显卡厂商Nvidia推出的计算平台)开发工具设计的量子化学计算和分子动力学(MD)模拟的算法和程序,尤其对目前唯一完全使用GPU技术开发的量子化学计算软件TeraChem做了完备的介绍,包括算法、实现的细节和程序目前的功能.此外,本文还对GPU在计算化学上将会发挥的作用做出了极为乐观的展望.
GPU;CUDA;计算化学;分子动力学;TeraChem
1 引言
计算化学是基于计算科学的原理来解决化学问题的一个化学分支学科.计算科学和技术的任何进步,都会迅速应用于计算化学,为计算化学带来长足的进步.而敏锐的计算化学家们,也时刻关注着计算科学的每一个新进展,切盼这些进展能对使用计算机从微观上了解化学和生命现象带来突破和契机.无疑,自2007年起GPU在计算化学上的应用和导致的一些具有革命意义的进展(且该浪潮的势头还正方兴未艾)对任何与计算化学有关的学科的科学工作者,都是应该密切关注和深入了解的.概要介绍GPU用于科学计算的基本技术和原理,穷极搜罗至今有关的成果,条分缕析地归类分析,为计算化学家和关注并需要计算化学成果的非计算化学家提供一个引导性的材料,使他们能把握前进的脉搏,而争取迅速迎头赶上,并在其中争得一席之地,是写这篇综述的初衷.
在这篇综述中,第二节将简单介绍GPU以及它在数值计算中应用发展的几个阶段及加速在GPU上的计算软件开发的工具;第三节将逐一介绍迄今使用GPU改进量子化学计算算法和程序的努力和一个极有前景的完全基于GPU的量子化学计算软件TeraChem;第四节介绍使用GPU对MD算法的改进和迄今能在GPU上运行的MD程序.最后简单总结并展望基于GPU的计算化学算法和程序的辉煌前景.
2 GPU在数值计算中的应用和软件开发工具
价格合理的GPU(graphics processing units,图形处理器)最先是为能在计算机游戏中生动真切显示三维图形而设计的计算机硬件.为了更好地显示三维(3D)图形,GPU需要对每个要显示的画面进行大量的浮点运算.GPU最先的设计者们完全没有意识到,他们在每一个GPU中安排的数百个计算单元具有能够进行数量可观的并行运算的特征,使得GPU在今天成了科学计算的宠儿.相比而言,一直在计算机中担负计算使命的每一个CPU(central processing unit,中央处理器)到今天也最多只能有数个运算单元(多核).GPU的设计特性使得巨大数目的并行事件同时进入GPU的RAM(random-access memory,随机存取存储器),即相同操作能在GPU的内存不同部分执行.这属于SIMD(single instruction,multiple data,单指令多数据)类型.典型的GPU包含许多分列成组的计算单元,每组共享能快速进入的内存和一个指令单位.这些高度密集的计算单元需要更大的cache和控制单元.GPU在数值计算中表现出的优异性能与已经趋于平缓发展的CPU相比展现出了令人耳目一新的表现(见图1,数据1收集到2008年6月).而之后不到三年的时间(到2011年3月),Nvidia的GeForce GTX 590的GPU的浮点运算峰值和内存带宽就又冲到了2488.3 GFlop·s-1和2×163.87 GB·s-1.2
GPU结构的本身具备进入科学计算的基本需求.最早将GPU用在数值计算中的企图可以追溯到20年前.3尽管之后各种努力和尝试不绝如缕,4,5但很长时间以来一直没有突破性的进展,原因是缺少科学计算编程的有效工具.当时一般是用特定的汇编语言或图形取向的应用编程接口(API,application programming interface),如OpenGL或DirectX等编程,工作量之巨大让人望而生畏.值得一提的工作有最早的Yang等6使用OpenGL作为载体编制出程序,在GPU上进行了分子动力学的模拟计算;之后有Anderson等7在GPU上进行量子蒙特卡罗(QMC)计算的尝试.
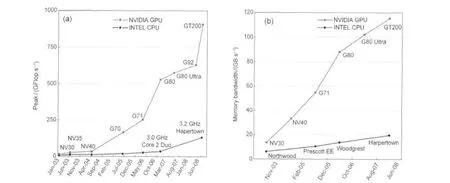
图1 GPU与CPU的浮点运算峰值和内存带宽的比较Fig.1 Comparisons of peak GFlop and memory bandwidth of GPU and CPUThe data come from NVIDIAwebsite.1GT200=GeForce GTX 280,G92=GeForce 9800 GTX,G80=GeForce 8800 GTX,G71=GeForce 7900 GTX, G70=GeForce 7800 GTX,NV40=GeForce 6800 Ultra,NV35=GeForce FX 5950 Ultra,NV30=GeForce FX 5800
由于GPU硬件快速发展的促动和科学计算高速化的迫切需求,在GPU上用于科学计算简易快捷的编程工具应运而生.它们是ATI Stream Technology8和Nvidia的CUDA(compute unified device architecture).9Stream计算机编程范式(paradigm)最先是ATI公司为在多个计算单元或多种计算硬件,如CPU和GPU间容易进行平行计算设计的,不需特别规定不同计算单元间的交流和传递.之后ATI公司在2006年被AMD公司收并,直到2010年后期,ATI品牌彻底被AMD取代.CUDA是一个类似于C语言的高级、直观的编程语言,并能被C语句调用. NVIDIA公司最近还推出了Parallel Nsight,9使CUDA能在微软公司最为普及的软件开发平台Visual Studio上使用.这惠及了数百万的软件开发工作者.可以毫不夸张地说,CUDA给GPU在科学计算上应用的飞速发展插上了翅膀.一时间诸多的与数值计算有关的科学家趋之若鹜,这已经在改变着科学计算的面貌了.
现有数值计算的程序编码已是浩如烟海,无论如何方便的在GPU上的编程工具,如CUDA,仍无法重复所有已有的编码和算法.若有简便的工具将现有的数值计算的程序转换到GPU上,将善莫大焉.在这个方向上的尝试和进展包括能在GPU上计算的量子化学和固体物理中常用的一些算法中的子程序库,如进行傅里叶变换的CUFFT10和线性代数库CUBLAS11和MAGMA.12
一开始影响GPU在科学计算上普及使用的另一障碍是早期GPU较低的数值精度,随着NVIDIA公司的支持32位浮点运算精度的G80系列GPU卡的推出和64位的GeForce 8800 GTX GPU卡的推出,用于科学计算的精度问题得到了彻底的解决.尽管在64位浮点运算精度下,运算速度会有所损失.
3 使用GPU的量子化学计算方法和程序
量子化学计算的基本方法,从头算(ab initio)和密度泛函理论(DFT)的最主要计算工作,是求得双电子排斥积分(也包括DFT中的交换-相关积分的数值求取)和生成Fock矩阵以及矩阵的对角化.
Yasuda13第一个认识到可使用GPU来提高计算电子排斥积分(ERI)的速度.他使用GeForce 8800 GTX GPU卡,发展出了使用GPU计算DFT的KS矩阵的Coulomb积分的算法,但仅适用于s和p基函数.这个算法在Taxol和Valinomycin的小基组和LSDA DFT计算里,使用GPU计算Coulomb势,仅需CPU常规计算时间的1/3.尽管使用的GPU仅支持单精度浮点数运算,但仍然有可以接受的精度. Yasuda的算法是在Gaussian 03程序14上完成的.
GPU对积分的计算只能包括s和p基函数是远远不够的.将GPU上的积分计算扩展到d及更高角量子数的基函数的困难在于目前最大的单个GPU也无法容纳这些高角量子数的计算壳层.如对于一个(ff|ff)双电子积分的壳层,在中间计算时共需要5376个浮点数,它们要做多次的乘法运算,最终需约104个浮点数.对于双精度,这相应于123008 bytes,远远超出了目前最大单片GPU的容量.有人在等待着很快有更大的GPU上市,但Asadchev等15另辟蹊径,近来提出了一个使用CUDA能计算到g型基函数的算法.他们将计算的中间结果存放在装置的内存,而不是GPU中,并将这些壳层块的积分重新排序,满足这些积分的频繁调用.这样做带来的问题是为了适应所有积分类型的有效排列的方式,原来在CPU下使用的大量程序需要重写.他们采用了由一个个模块自动生成的方式来解决这个问题.使用GTX275和Tesla T10 GPU卡,他们与GAMESS16比较了双电子排斥积分的运算速度. CPU能达到1 GFlop·s-1(每秒钟进行1 giga浮点运算),而GPU在单精度和双精度运算时能分别达到50和25 GFlop·s-1.
对DFTGGA中的XC(交换-相关)势的数值计算,仍是Yasuda17首先提出了在GPU上计算的算法.他采用的策略是在仅将XC较费时的步骤交由GPU,其余仍由CPU处理.使用NVIDIA的GeForce 8800 GTX卡,与2.8 GHz的Intel Pentium 4相比,该步计算的加速能达到40倍.
Aspuru-Guzik等18,19将开发基于GPU的量子化学算法的注意力放在了包括相关能校正的RI-MP2 (RI:resolution-of-the identity)方法上.除了对于含过渡金属的体系,MP2计算的平衡几何构型优于广为使用的DFT.MP2计算中最为耗时的是原子轨道基的积分向分子轨道基的变换.这一般是体系基函数数目N的5次方(O(N5)).引入RI积分近似可减少计算时间而又没有明显的精度损失.Aspuru-Guzik等分析RI-MP2计算各个步骤对计算机时间的需求,认识到RI-MP2计算中主要的时间消耗是在从半变换的三指标积分变换为分子轨道积分时的矩阵相乘.他们使用CUBLAS16的函数在GPU上执行矩阵相乘的运算(将CUBLAS的函数引入量子化学计算这还是首次).据他们自己的测试,在单精度下,计算速度能达到原来使用CPU的4.3倍.18使用NVIDIA Tesla C1060 GPU和4 GB的内存,他们测试了有168个原子的分子Valinomycin,在单精度、混合精度和双精度下分别能比CPU提速13.8、10.0和7.8倍,在双精度下,误差接近于零.他们的计算是植根于另一广为使用的商品化量子化学计算程序Q-Chem.20
在此还应提及一个不被化学家熟悉的、使用Daubechies波,而不是Gaussian型的基函数的程序包BigDFT.21BigDFT使用CUDA在GPU上编程,达到了极高的并行效率,使用GPU的版本能比CPU的提速达6倍.
量子化学计算程序Firefly22的前身是著名的量子化学程序GAMESS16的变种PC GAMESS.近来Firefly在它的MP4(SDTQ)计算中使用了GPU,对于Cl-(HF)5分子使用6-311+G(2d,2p)基组进行单点计算,在使用四个GPU(2 GTX295+2 C1060)时,计算速度提高了近10倍.23
通用的量子化学程序包Q-Chem20的一个突出特点是具有运算速度非常快的DFT计算功能.近来Q-Chem的开发者们Gan等24也将GPU用于进一步提高DFT计算速度.结合已有的可用于DFT一些步骤的GPU算法和Q-Chem在DFT计算上的改进,提出一个方案(task based programming,object-oriented design,OPENMP for multithreading,MPI for cross node communications,即基于作业编程、目标取向设计、用OPENMP作多线程和用MPI处理节点间的交通).作者自己的测试计算对Taxol分子(C47H51NO14),使用一片C1060 GPU,计算速度提高了24倍,若使用4片GPU,速度提高达到了102倍.
对将GPU使用到量子化学计算极为关注和付出努力的还有美国Iowa州立大学的Gordon教授. Gordon教授的研究小组是GAMESS程序16的维护者,并不断开发和发展.最先对GPU计算精度的不足曾影响了Gordon教授对之的投入,随着双精度的GPU卡的出现,Gordon教授和GAMESS研发小组变成了积极的参与者.Gordon教授和他的研发小组正在重写GAMESS中的程序,使之能在GPU上高效使用,相信很快就有这样的GAMESS版本推出.25
以上提到的近几年提出的使用GPU提高量子化学计算速度的算法中,除了BigDFT,21其余都是利用现有软件包,改善其中部分算法,在某一特定计算步骤,使计算速度得到提高.而刚于2010年推出的TeraChem程序则完全使用CUDA概念,重写了GPU上的量化计算的基本程序.26TeraChem是由美国Stanford大学的Ufimtsev和Martinez编写完成的,由美国PetaChem公司于2010年推出商业化版本.27由于他们在GPU的量子化学程序的先驱性的工作和突出贡献,Martinez教授被Gordon称为美国量子化学家的GPU的领头人.25
Ufimtsev和Martinez28提出的计算双电子排斥积分基于McMurchie-Davidson框架,检验了三种分配GPU/CPU工作和处理任务分块的模式(1B1CI、1T1CI和1T1PI),先进行由H原子组成的体系的(ss|ss)型积分的运算,然后推广到包括由p函数组成的基的排斥积分.他们发现若从Gaussian基的双电子排斥积分直接进入Fock矩阵,而不是如传统的将Gaussian轨道基的排斥积分收缩到Slater轨道(STO),在GPU下能有极高的效率.对于由64个H组成的体系,使用6-311G和三种模式计算排斥积分,分别耗费7.086、0.675和0.428 s,加上用CPU的预计算的0.009 s和GPU-CPU转换的0.883 s,最快的1T1PI模式共需1.320 s.而使用GAMESS在单CPU下则需170.8 s.Ufimtsev和Martinez29随后报道了完全使用GPU进行Hartree-Fock计算的方法.基于以前提出的1T1PI处理双电子排斥积分的方法28,他们提出了Hartree-Fock的J矩阵和K矩阵在GPU下依据大小分块处理的算法,还研究了多GPU的并行化处理:每个GPU处理自己的J矩阵子块,计算K矩阵时每个GPU处理K矩阵的不同的行.使用三个GPU卡(GTX280)做检验,能达到一个GPU卡的2.0-2.8的效率.他们测试了从24个原子到453个原子组成的分子的Hartree-Fock单点能量计算.使用3-21G和6-31G基组以及一个GPU卡(GeForce 280 GTX),在双精度下,与GAMESS相比,速度能提高40到600多倍,而总能量的差别可忽略不计.
Ufimtsev和Martinez系列文章的第三篇26是关于能量解析梯度计算的GPU处理,包括Coulomb和交换部分计算的处理.加上多GPU卡的并行计算的处理,该方法可用于Hartree-Fock能量和DFT能量的解析梯度的计算.使用GPU程序和单个GPU卡(GeForce 295 GTX),与在4核2.66 GHz CPU上运行的GAMESS程序相比,从小到大的分子,GPU仍然可有数倍到超过百倍的速度提高.
这样,他们提出的在GPU上运行的量子化学计算的算法,涵盖了基本的从头算/DFT的全部三个耗时步骤(双电子积分、Hartree-Fock/Kohn-Shan矩阵处理和能量的解析梯度).基于此,Martinez推出了他们的TeraChem程序.262010年春推出的为1.05版,能进行闭壳层的Hartree-Fock和DFT的单点能量计算,基于解析梯度的几何构型极小化和过渡态优化的计算,以及基于以上计算(能量和解析梯度)的动力学模拟(MD)和简单的QM/MM计算.使用TeraChem,很容易检验和实现上述他们的三篇论文的结果.最近(2011年4月)推出的v1.45版已能进行包括d轨道和含过渡金属原子体系的HF和DFT计算,并能达到与sp轨道差不多的速度提高.具体算法的改进和实现还未见有详细报道.
TeraChem更为不凡,需引起密切关注的原因是这是第一个完全基于GPU,从零开始编制的量子化学计算程序.本文引述的在Ufimtsev和Martinez的工作之前或之后的关于GPU的量子化学计算的算法的文章,虽都基于某一特定量子化学程序包实现,但至今还都没有能进入公开发行的程序包中,有些或许只能永远停留在纸面上.而TeraChem一进入人们的视野,就以完备的商品软件的姿态出现.据PetaChem网站披露,TeraChem在2010年春天一推出,短短半年就有超过4000份的试用版本发出,展现出蓬勃的生命力.
量子蒙特卡罗(QMC)是求解与时间无关的薛定谔方程的精确方法之一,30并且QMC很容易并行化.Anderson等7在2007年就提出了在GPU上加速QMC计算的方案.使用CUDA核优化缓存使用,在GPU上进行需求很大的并行计算.对于8-32个原子的体系,比CPU上的执行,速度能有5倍的提高. Meredith等31在单精度的GPU上用量子簇近似,使用规整晶格上的二维Hubbard近似研究超导在临界温度下的混乱效应,在GPU上使用CUBLAS库执行矩阵乘法,得到与CPU上的双精度可比的结果.
4 分子动力学算法和程序中的GPU
MD计算对生物分子的模拟自McCammon等对酶的计算为肇基,32当时的计算水平是500个原子,模拟的时间10 ps.之后飞速发展,到了今天,模拟的生物体系包括数百万个原子,模拟时间能持续到微秒都已成常规.而科学家对生命现象的深入了解仍在向计算化学家不断提出更高的要求.如对蛋白质折叠的MD模拟研究,折叠行为约需毫秒来完成,如以纳秒为步长做模拟,且每步计算花费一天计,这样的模拟就需执行一百万天,即差不多3000年.这确实是对计算化学家提出了严峻的挑战.虽然有人花费巨资搭建豪华计算机系统,如日本的总耗资超过一千五百万美元的MDGRAPE-3系统,33,34这是使用专门设计的硬件加速搜寻表以加速直接空间非键的计算.但这样的耗费巨资并为某一特定问题专门设计的硬件完全无法做广泛的推广,且不说耗资惊人了.而GPU适时的出现为MD对生物的模拟计算带来了曙光.
由于在GPU下编程的困难,早期试图在GPU上提高MD计算速度的努力有许多是走入迷途的,报导的结果也是使人信疑参半.至今被普遍接受的GPU上MD算法的成功改进仍属本文开始提及的Yang等6的工作,尤其值得赞许的是该工作是在CUDA推出之前,直接使用OpenGL编程,在GPU上实现需惊人的工作量,并达到了10倍以上速度的提高.虽然他们的算法相当简捷,在模拟过程中仅包含Lennard-Jones相互作用和建立一个邻接目录,而缺乏对生物体系模拟的许多计算步骤,但这个先驱性的工作仍有力地证实了GPU能在MD模拟上发挥重要作用.
早期使用GPU改进MD运算速度的尝试大都局限于使用简化的模型,或者只针对于特定的问题做处理,而不是着眼于对真正MD的全面改进.Liu等35是第一个使用CUDA来对MD的计算进行改进的.他们仅在GPU上计算简化的范德华势,而避免了对凝聚态体系MD模拟的复杂的乘积运算.Stone等36在他们的文章里仔细讨论了一系列用于分子模拟的算法,提出了第一个直接空间范德华势和静电势在GPU上合并处理的方法,但该方法并不包括任何实际的MD计算,而仅仅是在被模拟体系临近库仑势的时间平均的计算中使用GPU,并引入到MD程序包NAMD37中,诞生了NAMD的GPU加速的新版.
之后还有一些使用GPU对MD算法的局部改进,38,39直到Anderson等40将共价项的贡献与使用GPU计算的范德瓦尔和简谐键势项加入到他们的HOOMD程序包41中,用于研究非离子液体.可以认为,GPU版的HOOMD朝向真正完整的GPU的MD程序前进了一大步.
真正能称之为使用GPU的MD程序产品是到2009/2010年才出现的.至今只有三个MD程序包可认为基本满足,它们分别是Harvey等42的ACEMD、Friedrichs等43的OpenMM和Philips等37的NAMD.
ACEMD可谓是第一个支持全部GPU加速功能的处理凝聚相的MD程序包,可处理周期性边界和短程、长程的静电作用.使用GPU加速后,有很明显的速度提高.OpenMM是一个程序库,起先仅是对于小的和中等大小的体系使用非键项的间接求和方法来计算溶剂的广义Born模型,之后Eastman和Pande44对之改进,显性计算进行溶剂的MD模拟.最新推出(2011年3月11日)的OpenMM版是v3.0.著名MD程序包GROMACS45新近的GPU版将OpenMM纳入其中,可在GPU上大大提高MD计算的速度.NAMD的v2.7b2版也添加了GPU加速的明确进行溶剂化计算的功能,在对一个由25095个原子组成的生物体系的模拟计算,速度提高到了8.6倍.NAMD最新的版本是v2.8b1,仍是以CPU计算为主,在多节点间进行MD计算,而将之置于GPU的环境,使用单片或多片GPU来提高CPU的单个节点内的计算效率以达到提高MD计算速度的目的.
AMBER程序可谓是MD计算机程序家族中最为著名的一员,46由一些使用AMBER力场47进行与MD有关计算的程序包组成.最近AMBER程序家族的PMEMD程序在显性溶剂PME(particle mesh Ewald)和广义Born模型(GB,generalized Born)的处理中使用Nvidia的GPU,并因之推出了AMBER11版,48但使用GPU后的速度提高和计算精度还都有待于检验.
最近推出的另一MD程序HOOMD-BLUE 0.9.241也声称支持GPU的运算.算法的作者声称他们的整个MD计算都在GPU上进行,单片GPU运算的速度相当于30个微处理器.40
尽管有诸多的对MD在GPU上应用的算法的创立和改进,和相应的能在GPU上运行的MD程序的推出,其中某些还声称是完全使用了在GPU上基于CUDA开发的新算法,但相对于GPU的量子化学计算程序能对速度有50-100倍的提高,GPU上的MD程序的速度提高还不尽如人意(最高速度提高的倍数大约为10,但据我们验证,除特殊情况,基本无法达到这个速率),这是由MD计算的本质决定的.尽管今天的CPU,甚至GPU的计算速度已足够快,但硬件间数据传递的速度仍是瓶颈,而MD计算却是非常依赖和需要这些传输.GPU运算速度的提高无助于这些传输,甚至加重了节点间数据传输的压力.这就是为什么至今GPU上的MD程序仅限于单个GPU(OpenMM和AMBER)或单个节点内的多个GPU(ACEMD).仅NAMD试图在多节点上应用GPU改进MD的计算速度,试图模拟非常大的体系,而不是优化单节点内的运行,这需要数量众多的GPU卡,而仅有差强人意的速度提高,且“性价比”还相当低.
5 结语
近年由CUDA带动的GPU在科学计算上的蓬勃应用和发展,不可能不进入计算化学领域.自2010年以来,接连有GPU在计算化学(包括量子化学计算和MD模拟)的综述文章发表,如Götz等49题为“Quantum Chemistry on GPU”的综述,和Xu等50对GPU在生物分子MD模拟的进展的综述(本文的一些材料间接引自这两篇综述),最近美国Chemical&Engineering News上的一篇题为“GPU革命(The GPU Revolution)”的短文也对GPU将对计算化学的贡献和前景给出了极为乐观的评价和期许.25
纵观由三年来多方努力和如雨后春笋般的在GPU上的量子化学计算和MD模拟算法和程序的呈现,仅有Ufimtsev和Martinez辛勤创立的Tera-Chem软件是完全从零的基础上、完全为GPU计算设计和实现,却也达到了计算速度的可喜提高,而其它量子化学程序和差不多所有的MD程序都还有各种各样的缺憾和不足.TeraChem或许会成为量子化学计算的一个新里程碑.如同四十年前Gaussian 70和九十年代初量子化学软件的商品化大潮,呈现以二十年为周期的飞跃.
同时,Nvidia会不断推出容量更大、速度更快的GPU,而价格也会愈来愈低,CUDA也会不断扩充新功能,并愈来愈完善,使得在GPU上编制科学计算程序成为常规.而AMD的ATI Stream Technology和相应的硬件或许在很近的将来会成为Nvidia的GPU卡和CUDA的有力竞争者,而且GPU在科学计算上的应用也将出现百花齐放的繁盛局面.这些都使我们相信,我们处在一个计算化学新时代来临的前夜.
(1)NVIDIACUDA.Compute Unified DeviceArchitecture Programming Guide Version 3.0.http://www.nvidia.com/object/ cuda_develop.html(accessed March 6,2010).
(2) Comparison of Nvidia Graphics Processing Units.http://en. wikipedia.org/wiki/Comparison_of_Nvidia_graphics_ processing_units(accessed March 6,2010).
(3) Lengyel,J.;Reichert,M.;Donald,B.R.;Greenberg,D.P. Comput.Graph.1990,24,327.
(4) Bohn,C.A.Joint Conference on Intelligent Systems 1999(JCISʹ 98)1998,2,64.
(5) Hoff,K.E.,II.;Culver,T.;Keyser,J.;Ming,L.;Manocha,D. Fast Computation of Generalized Voronoi Diagrams Using Graphics hardware.In Proceeding of SIGGRAPH 99,Danvers, August 8-13,1999;Assison-Wssley Publishing Company, 1999,277-286.
(6)Yang,J.;Wang,Y.;Chen,Y.J.Comput.Phys.2007,221,799.
(7)Anderson,A.G.;Goddard,W.A.,III.;Schroder,P.Comput. Phys.Commun.2007,177,265.
(8)ATI Stream Technology,http://www.amd.com/US/PRODUCTS/ TECHNOLOGIES/STREAM-TECHNOLOGY/Pages/ stream-technology.aspx(AccessedApril 13,2011).
(9) CUDA:Santa Clara,CA.http://www.nvidia.com/object/ cuda_home_new.html(accessedApril 13,2011).
(10) NVIDIA:Santa Clara,CA,CUFFT Library.http://developer. download.nvidia.com/compute/cuda/2_3/toolkit/docs/ CUFFT_Library_2.3.pdf(accessed March 6,2010).
(11) NVIDIA:Santa Clara,CA,CUBLAS Library 2.0.http:// developer.download.nvidia.com/compute/cuda/2_0/docs/ CUBLAS_Library_2.0.pdf(accessed March 6,2010).
(12) Innovative Computing Laboratory,University of Tennessee, MatrixAlgebra on GPU and MulticoreArchitectures.http://icl. cs.utk.edu/magma(accessed March 6,2010).
(13)Yasuda,K.J.Comput.Chem.2008,29,334.
(14) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, Revision B.01;Gaussian Inc.:Pittsburgh,PA,2003.
(15)Asadchev,A.;Allada,V.;Felder,J.;Bode,B.M.;Gordon,M.S.; Windus,T.L.J.Chem.Theory Comput.2010,6(3),696.
(16) (a)Schmidt,M.W.;Baldridge,K.K.;Boatz,J.A.;Elbert,S.T.; Gordon,M.S.;Jensen,J.H.;Koseki,S.;Matsunaga,N.; Nguyen,K.A.;Su,S.;Windus,T.L.;Dupuis,M.;Montgomery, J.A.J.Comput.Chem.1993,14,1347. (b)Gordon,M.S.;Schmidt,M.W.Advances in Electronic Structure Theory:In Theory and Applications of Computational Chemistry:the First Forty Years;Dykstra,C.E.,Frenking,G., Kim,K.S.,Scuseria,G.E.,Eds.;Elsevier:Amsterdam,2005; p 1167.
(17)Yasuda,K.J.Chem.Theory Comput.2008,4,1230.
(18)Kermes,S.;Olivares-Amaya,R.;Vogt,L.;Shao,Y.; Amador-Bedolla,C.;Aspuru-Guzik,A.J.Phys.Chem.A 2008, 112,2049.
(19) Olivares-Amaya,R.;Watson,M.A.;Edgar,R.G.;Vogt,L.; Shao,Y.;Aspuru-Guzik,A.J.Chem.Theory Comput.2010,6, 135.
(20) Shao,Y.H.;Fusti-Molnar,L.;Jung,Y.S.et al.Phys.Chem. Chem.Phys.2006,8,3172.
(21) Genovese,L.;Ospici,M.;Deutsch,T.;Mehaut,J.F.;Neelov,A.; Goedecker,S.J.Chem.Phys.2009,131,34103.
(22) Granovsky,A.A.Firefly version 7.1.G.http://classic.chem.msu. su/gran/firefly/index.html(accessedApril 4,2011).
(23) http://classic.chem.msu.su/gran/gamess/cuding.html(accessed April 4,2011).
(24) Gan,Z.;Shao,Y.;Kong,J.;Olivares-Amaya,R.;Aspuru-Guzik, A.http://www.nvidia.com/content/GTC/documents/ 1050_GTC09.pdf(accessedApril 4,2011).
(25) Wolf,L.Chemical and Engineering News 2010,88,27.
(26) Ufimtsev,I.S.;Martinez,T.J.J.Chem.Theory Comput.2009, 5,2619.
(27)TeraChem.http://www.petachem.com(accessed March 6,2010).
(28) Ufimtsev,I.S.;Martinez,T.J.J.Chem.Theory Comput.2008, 4,222.
(29) Ufimtsev,I.S.;Martinez,T.J.J.Chem.Theory Comput.2009, 5,1004.
(30) Ceperley,D.;Alder,B.Quantum Monte Carlo.Science 1986, 231,555.
(31) Meredith,J.S.;Alvarez,G.;Maier,T.A.;Schulthess,T.C.; Vette,J.S.Parallel Comput.2009,35,151.
(32)McCammon,J.A.;Gelin,B.R.;Karplus,M.Nature 1977,267, 585
(33) Susukita,R.;Ebisuzaki,T.;Elmegreen,B.G.;Furusawa,H.; Kato,K.;Kawai,A.;Kobayashi,Y.;Koishi,T.;McNiven,G. D.;Narumi,T.;Yasuoka,K.Comput.Phys.Commun.2003, 155,115.
(34) Narumi,T.;Ohno,Y.;Noriyuk,F.;Okimoto,N.;Suenaga,A.; Yanai,R.;Taiji,M.In From Computational Biophysics to Systems Biology:MDGRAPE-3;Meinke,J.,Zimmermann,O., Mohanty,S.,Hansmann,U.H.E.Eds.;J.von Neumann Institute for Computing:Jülich,2006;p 29.
(35) Liu,W.;Schmidt,B.;Voss,G.;Müller-Wittig,W.In High Performance Computing-HiPC 2007:Lecture Notes in Computer Science;Aluru,S.,Parashar,M.,Badrinath,R., Prasanna,V.K.Eds.;Springer,Berlin/Heidelberg,2007; Vol.4873,p 185.
(36) Stone,J.E.;Phillips,J.C.;Freddolino,P.L.;Hardy,D.J.; Trabuco,L.G.;Schulten,K.J.Comput.Chem.2007,28,2618.
(37) Phillips,J.C.;Stone,J.E.;Schulten,K.Adapting a Message-Driven ParallelApplication to GPUAccelerated Clusters.In SCʹ08:Proceedings of the 2008 ACM/IEEE conference on Super Computing,1-9,IEEE Press,Piscataway, NJ,USA,2008.
(38) van Meel,J.A.;Arnold,A.;Frenkel,D.;Portegies Zwart,S.F.; Belleman,R.G.Mol.Simulat.2008,34,259.
(39) Rapaport,D.C.Comput.Phys.Commun.2011,182,926.
(40)Anderson,J.A.;Lorenz,C.D.;Travesset,A.J.Comput.Phys. 2008,227,5342.
(41)HOOMD:General Purpose Molecular Dynamics on Multiple GPUs Implemented Using CUDAJoshuaA.Anderson Path to Petascale:Adapting GEO/CHEM/ASTROApplications for Accelerators andAccelerator Clusters,April 2009.http://www. ncsa.uiuc.edu/Conferences/accelerators/agenda.html(accessed April 4,2011).
(42) Harvey,M.J.;Giupponi,G.;De Fabritiis,G.J.Chem.Theory Comput.2009,5,1632.
(43) Friedrichs,M.S.;Eastman,P.;Vaidyanathan,V.;Houston,M.; Le Grand,S.;Beberg,A.L.;Ensign,D.L.;Bruns,C.M.; Pande,V.S.J.Comput.Chem.2009,30,864.
(44) Eastman,P.;Pande,V.S.J.Comput.Chem.2010,31,1268.
(45)GROMACS http://www.gromacs.org(accessedApril 4,2011).
(46) Pearlman,D.A.;Case,D.A.;Caldwell,J.W.;Ross,W.S.; Cheatham,T.E.,III.;DeBolt,S.,Ferguson,D.;Seibel,G.; Kollman,P.Comp.Phys.Commun.2005,91,1.
(47) Cornell,W.D.;Cieplak,P.;Bayly,C.I.;Gould,I.R.;Merz,K. M.,Jr.;Ferguson,D.M.;Spellmeyer,D.C.;Fox,T.;Caldwell, J.W.;Kollman,P.A.J.Am.Chem.Soc.1995,117,5179.
(48) Case,D.A.;Darden,T.A.;Cheatham,T.E.,III.;Simmerling,C. L.;Wang,J.;Duke,R.E.;Luo,R.;Walker,R.C.;Zhang,W.; Merz,K.M.;Roberts,B.;Wang,B.;Hayik,S.;Roitberg,A.; Seabra,G.;Kolossváry,I.;Wong,K.F.;Paesani,F.;Vanicek,J.; Liu,J.;Wu,X.;Brozell,S.R.;Steinbrecher,T.;Gohlke,H.;Cai, Q.;Ye,X.;Wang,J.;Hsieh,M.J.;Cui,G.;Roe,D.R.; Mathews,D.H.;Seetin,M.G.;Sagui,C.;Babin,V.;Luchko, T.;Gusarov,S.;Kovalenko,A.;Kollman,P.A.AMBER 11; University of California:San Francisco.
(49) Götz,A.W.;Wölfle,T.;Walker,R.C.Annual Reports in Computational Chemistry;Elsevier:Amsterdam,2010;Vol.6, p 21.
(50) Xu,D.;Williamson,M.J.;Walker,R.C.Annual Reports in Computational Chemistry;Elsevier:Amsterdam,2010;Vol.6, p 1
April 15,2011;Revised:June 9,2011;Published on Web:June 24,2011.
GPU Triggered Revolution in Computational Chemistry
BAO Jian-Zhang1FENG Xin-Tian2YU Jian-Guo2,*
(1Department of Systems Science,School of Management,Beijing Normal University,Beijing 100875,P.R.China;2College of Chemistry,Beijing Normal University,Beijing 100875,P.R.China)
Over the last 3 years,the use of graphics processing units(GPU)in general purpose computing has been increasing because of the development of GPU hardware and programming tools such as CUDA(compute unified device architecture).Here,we summarize the progress in algorithms and the corresponding software with regard to computational chemistry using GPU including quantum chemistry and molecular dynamics simulations in detail.We introduce and explore the newly developed TeraChem program,which is unique quantum chemical software and we discuss the algorithms, implementations,and functionality of the program.Finally,we give an optimistic outlook for the use of GPU in computational chemistry.
GPU;CUDA;Computational chemistry;Molecular dynamics;TeraChem
∗Corresponding author.Email:jianguo_yu@bnu.edu.cn;Tel:+86-10-58802051.
The project was supported by the National Natural Science Foundation of China(20733002,20873008,21073014).
国家自然科学基金(20733002,20873008,21073014)资助项目
O641
猜你喜欢
中学生数理化(高中版.高考数学)(2022年1期)2022-04-26
数学小灵通(1-2年级)(2020年6期)2020-06-24
人大建设(2019年12期)2019-05-21
学周刊(2019年3期)2019-01-11
瞭望东方周刊(2017年42期)2017-12-05
环球时报(2017-03-30)2017-03-30
中学生数理化·八年级数学人教版(2017年2期)2017-03-25
中国卫生(2015年3期)2015-11-19
中国校外教育(下旬)(2015年3期)2015-05-22
