
应用重叠延伸法合成人源明胶基因及其克隆
2011-11-26 00:30段慧明陈劲春
湖南师范大学自然科学学报 2011年3期
段慧明,陈劲春
(北京化工大学生命科学与技术学院,北京 100029)
明胶是一种从动物(牛、马 猪)的胶原结缔组织和皮经过一系列化学处理以后所得到的蛋白质.干燥后的明胶涂层呈透明状,具有一定的机械强度.在感光工业, 医药工业、食品工业以及其他工业中获得非常广泛的应用.明胶因使用原料、生产方式和质量不同又分为食用明胶、医用明胶、工业明胶、骨胶和照相用胶等.明胶可以从许多不同来源的胶原蛋白制取.牛骨、皮、猪皮和鱼是主要的商业来源.因此,它可能来源于农业或者非农业.明胶也用作啤酒的澄清剂和很多产品的稳定剂、增稠剂和组织形成剂.
目前国内还没有表达重组明胶的相关报道,国内生产明胶都是通过物理化学方法提取获得.制造明胶的过程包括从动物组织提取胶原并将其转化成明胶.这个过程产生大小和电荷均不同的多肽异源混合物.使用微生物系统制造明胶是另一种策略.这种策略包括通过表达具有特定长度和组成的胶原基因片段直接产生明胶.使用这些胶原片段的研究表明它们能被用作明胶的许多医学用途的动物源明胶的替代品[1].
1 材料与方法
1.1 材料
大肠杆菌DH5α由本实验室保存,克隆质粒pGEM-T、T4DNA连接酶购自Promega公司,pfu、Taq酶、限制性内切酶HindⅢ、SnabⅠ、SalⅠ购自Takara公司.玻璃奶购自博大泰克公司,DNAMarker购自华美公司.其余试剂为国产分析纯.PCR引物由Invitrogen 公司合成,PCR反应在PERKIN ELMER公司2400型扩增仪上进行.
培养基:LB、LB固体平板按英骏公司的方法配制.
1.2 分子克隆常规操作
质粒提取、PCR产物回收参照分子克隆实验手册进行.
1.3 寡核苷酸片段的合成
参照人Ⅰ型胶原蛋白α1链结构基因的序列选取了α1链第531~630个氨基酸的编码序列设计人工合成该基因的方案.并且首先将该基因的序列进行遗传密码的优化设计,即根据密码子使用偏好把人的编码方式改变为毕赤酵母的编码方式,将编码胶原蛋白的基因分成10段,相邻两个片段之间的互补序列长度为17~20个碱基.在该基因的5'端引入了SnabⅠ酶切位点,在3'端引入了HindⅢ酶切位点.全基因序列的划分情况如表1所示(带下划线处均为重叠区), 寡核苷酸片段的合成由北京英骏公司完成.

表1 化学合成人源明胶基因的寡核苷酸片段
1.4 寡核苷酸片段的连接和组装
采用重叠延伸PCR技术,按如下方法将寡核苷酸片段连接成一完整人源明胶基因:①将明胶基因片段1~10分别重叠延伸形成寡核苷酸片段(1~4)、(5~8)、(9~10),PCR反应体系为0.2 μL dNTP(25 mmol/L)、0.2 μL pfu酶、2 μL 10×Buffer、寡核苷酸片段、纯净水16.8 μL,总反应体系为20 μL.PCR反应条件:94 ℃预变性5 min→[94 ℃,1 min→64 ℃ 1 min→72 ℃ 1 min] ×25个循环→72 ℃ 10 min→4 ℃;②将步骤①得到的PCR产物回收后按(1~4)→(5~8)连接,反应体系为0.2 μ LdNTP(25mmol/L),引物1和引物8各0.5 μL, 2 μL 10×Buffer, (1~4) 和 (5~8)回收片段各0.5 μL, 0.2 μLpfu酶, 纯净水15.6 μL,总反应体系为20 μL.PCR反应条件:94 ℃预变性5 min→[94 ℃,1 min→55 ℃ 1 min→72 ℃ 5 min] ×1个循环(获得1~8模板),暂停加引物1和8各0.5 μL→94 ℃预变性5 min →[94 ℃,1 min→64 ℃ 1 min→72 ℃ 1 min] ×25个循环→72 ℃ 10 min→4 ℃;③将步骤②得到的PCR产物回收后按(1~8)→(9~10)连接,反应体系为0.2 μL dNTP(25 mmol/L),引物1和引物10各0.5 μL, 2 μL 10×Buffer, (1~8) 和 (9~10) 回收片段各0.5 μL, 0.2 μL pfu酶, 纯净水15.6 μL, 总反应体系为20 μL ,PCR反应条件同步骤2.至此,得到全基因.
1.5 人源明胶基因的克隆
以1.4中得到的(1~10)为模板, 用引物1和引物10扩增得到全基因,PCR反应体系为:0.5 μL dNTP(25 mmol/L)、0.5 μL Taq酶、1 μL(1~10)模板、引物1和引物10各1 μL,5 μL 10×Buffer,41 μL 水,总反应体系为50 μL,PCR反应条件同上.PCR反应后走15 g/L琼脂糖凝胶电泳, 检测扩增片段的大小和特异性, 将目的片段切下, 用玻璃奶法回收纯化目的片段.
1.6 目的片段的克隆及重组子筛选
将回收的目的片段克隆到Promega公司的pGEM-T载体上,转化大肠杆菌DH5α,涂布于含有氨苄青霉素的平板上,培养过夜,挑取菌落接种培养,用碱变性法进行质粒DNA的小量提取,酶切鉴定.
1.7 DNA序列测定
将鉴定正确的菌液送英骏公司对扩增的目的基因片段测序确定碱基序列.
2 结果
2.1 组装与克隆
组装与克隆步骤如图1所示.
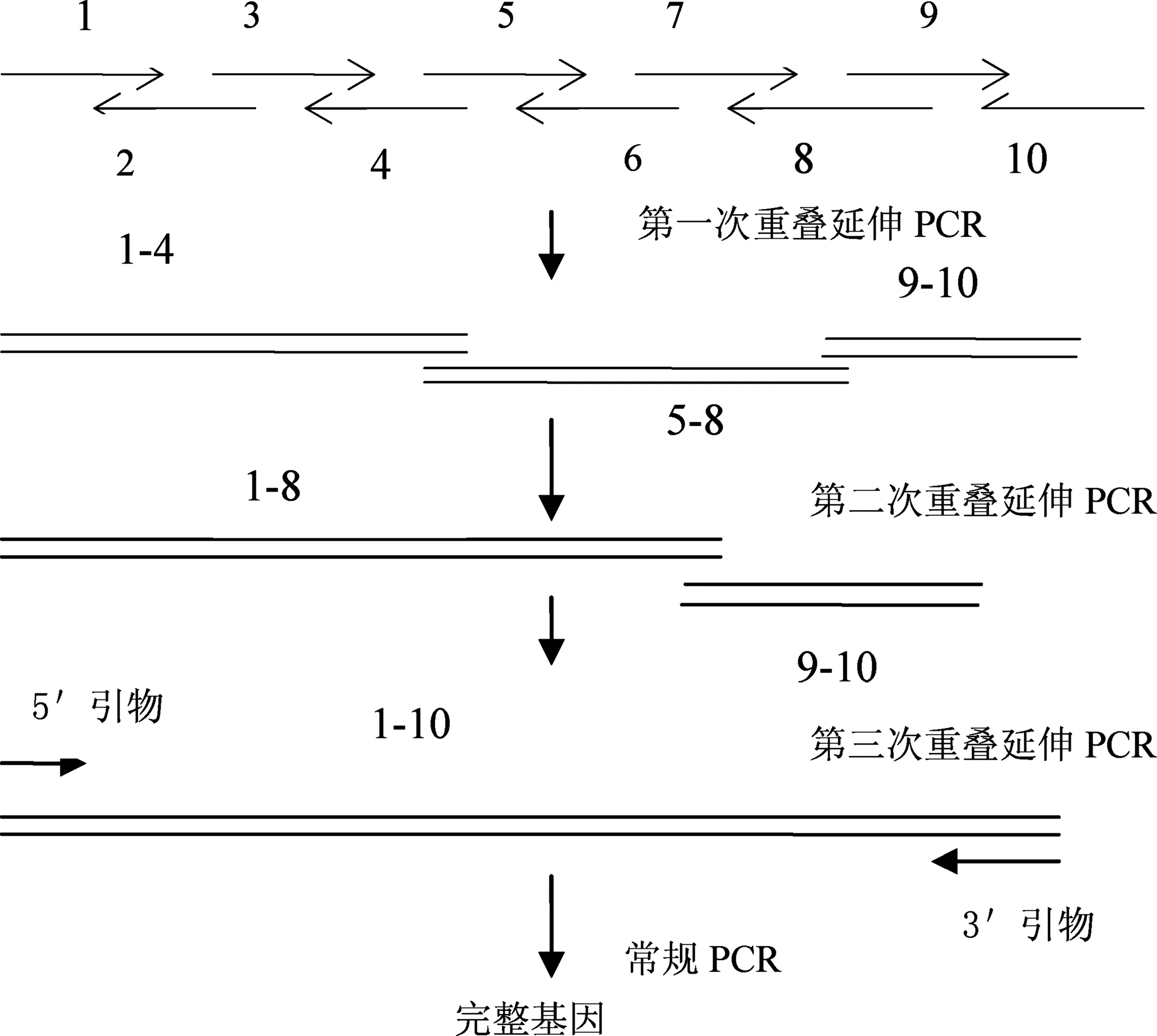
图1 化学合成基因流程图
2.2 PCR 扩增及其产物的电泳鉴定结果
以PCR产物1~10为模板,引物1和10分别为上游和下游引物进行PCR扩增,扩增条件见方法1.4,扩增产物进行15 g/L琼脂糖凝胶电泳,电泳结果见图2.
2.3 人源明胶基因的全序列测定
经过测序,序列与理论序列一致,该基因全长300 bp,带下划线处分别为SnabⅠ、HindⅢ酶切位点.
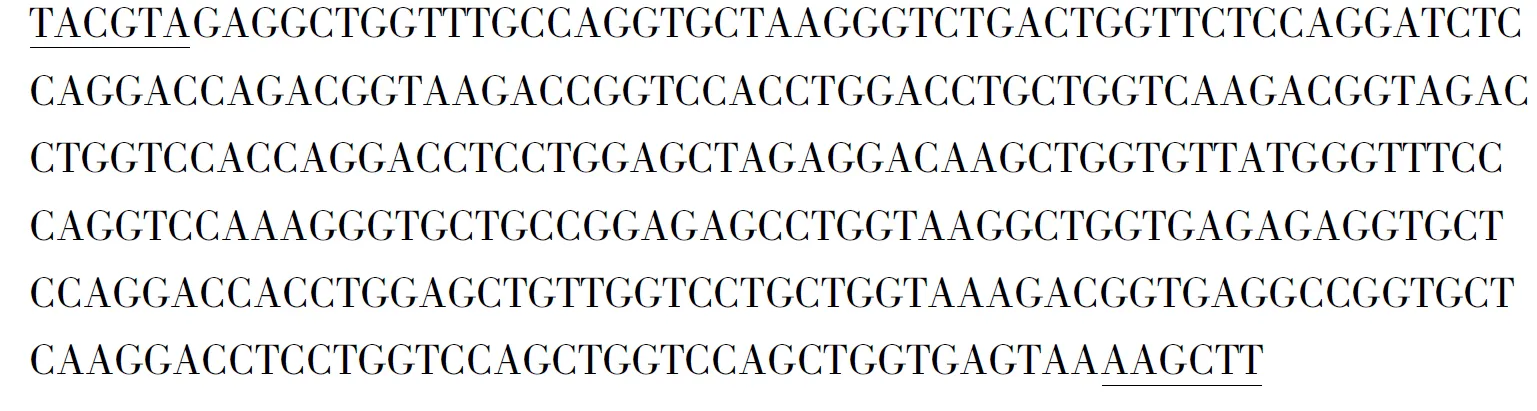

1:明胶基因PCR扩增产物;M:100 bp DNA plus图2 目的基因PCR扩增产物
3 讨论
胶原蛋白的结构基因由于具有三联体Gly-X-Y结构,重复序列较高,采用传统的磷酸化补齐方法很难获得目的基因,而重叠延伸PCR技术(gene splicing by overlap extension PCR,简称SOE PCR)[2-3]由于采用具有互补末端的引物,使PCR产物形成了重叠链,从而在随后的扩增反应中通过重叠链的延伸,将不同来源的扩增片段重叠拼接起来. 其原理是: 使用2对引物P1,P2和P3,P4经过第一轮PCR分别扩增出2个DNA片段A和B,其中P2和P3为内引物,具有部分序列重叠互补,使A的3′端和B的5′端具有同源互补序列; 第二轮PCR再以A和B混合作为模板,经变性,退火,复性时,A的正链和B的负链的3′端同源互补序列将结合在一起,使两条链互为引物,通过PCR得到延伸和扩增,从而连接A和B. 其特点是不需要使用限制性内切酶和连接酶,可以避免引入限制性酶切位点的核苷酸序列,可将2个DNA片段精确地连接在一起. 使用这种方法时,为了尽可能避免或减少碱基错配,往往需加大模板、引物和dNTP的用量,同时减少PCR的循环数,因此有时在其PCR产物的凝胶电泳图像中仍可见模板DNA的相应条带.因此应用SOE时,每一轮PCR产物最好经电泳分离后切胶回收纯化以除去多余的引物和其他非特异序列.此技术利用PCR技术能够在体外进行有效的基因重组,而且不需要内切酶消化和连接酶处理,可利用这一技术很快获得其他依靠限制性内切酶消化的方法难以得到的产物.重叠延伸PCR技术成功的关键是重叠互补引物的设计.重叠延伸PCR在基因的定点突变、融合基因的构建、长片段基因的合成、基因敲除以及目的基因的扩增等方面有广泛而独特的应用.
参考文献:
[1] 彭必先, 陈丽娟.从胶原到明胶[J]. 明胶科学与技术, 1994, 14(2):57.
[2] MULLINAX R L, GROSS E A, HAY B N,etal. Expression of a heterodimeric fab antibody protein in one cloning step[J]. Biotechniques, 1992, 12(6):864-869.
[3] HORTON R M, HUNT H D, HO S N,etal. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension[J]. Gene, 1989,77(1):61-68.
[4] WARRENS A N, JONES M D, LECHLER R I. Splicing by overlap extension by PCR using asymmetric amplification: an improved technique for the generation of hybrid proteins of immunological interest[J]. Gene, 1997,186(1):29-35.
[5] J·萨姆布鲁克, E·F·弗里奇, T·曼尼阿蒂斯. 分子克隆实验指南[M].金冬雁,黎孟枫,译.北京:科学出版社,1992.
[6] 胡显文,高丽华.炭疸毒素受体ATR cDNA的合成和克隆及ATR-Fc融合蛋白的真核表达载体的构建[J].中国生物工程杂志,2005,25(12):1-8.
[7] 陈红英,崔保安,李新生,等.融合基因Mchil-18/Mchifn-α克隆及其原核表达载体的构建[J].农业生物技术学报,2007, 15(3):388-392.
猜你喜欢
化工设计通讯(2022年6期)2023-01-02
南昌大学学报(理科版)(2021年3期)2021-10-13
现代畜牧科技(2021年8期)2021-10-13
生物工程学报(2018年5期)2018-06-11
癌症进展(2016年10期)2016-03-20
中国组织化学与细胞化学杂志(2016年3期)2016-02-27
中国生化药物杂志(2015年7期)2015-07-07
食品工业科技(2014年13期)2014-03-11
实验动物与比较医学(2014年4期)2014-02-28
河南科技(2014年22期)2014-02-27
