
5-氮-2'脱氧胞苷对Reh细胞增殖及RUNX3基因启动子区甲基化状态的影响
2011-08-07 06:21吴明彩李大彩毕富勇
皖南医学院学报 2011年6期
吴明彩,蒋 明,李大彩,毕富勇
(1.皖南医学院 生物化学教研室,安徽 芜湖 241002;2.第二炮兵芜湖鸠江干休所 卫生所,安徽 芜湖 241000)
DNA甲基化被认为是肿瘤形成的重要机制之一,抑癌基因可因高度甲基化而失活,这种基因沉默调控与体细胞突变共同促进了肿瘤的发展[1]。人类RUNX3(runt-related transcription factor 3)是一种新型的抑癌基因,位于1号染色体的1p36.1[2],已在人类多种实体瘤中发现存在RUNX3启动子区的异常甲基化,而急性白血病中该基因的甲基化状态的研究尚处于探索阶段。本实验选用白血病Reh细胞株为研究对象,进行体外培养,采用MTT、MSP以及RT-PCR方法检测5-氮-2'脱氧胞苷对Reh细胞生长增殖、RUNX3基因启动子区甲基化状态及表达的影响,探讨去甲基化制剂对Reh细胞增殖及其RUNX3基因表达的调控作用。
1 材料与方法
1.1 材料
1.1.1 主要试剂与药品 RPMI 1640培养基(上海生工);胎牛血清(BIO CHROM);DNA提取试剂盒(Promega公司);DNA甲基化试剂盒(Qiagen公司);Trizol试剂(上海生工);PCR试剂盒(Promega公司);RT-PCR(Promega公司);DNA Marker(上海生工);琼脂糖(西班牙进口);DNaseⅠ(上海生工)。引物由上海生工公司合成。
1.1.2 细胞培养 人白血病细胞系Reh购自中科院上海细胞库(ATCC建系)。Reh细胞于 RPMI 1640培养基(含10%胎牛血清),37℃、5%CO2、饱和湿度CO2培养箱中孵育,倒置显微镜下观察细胞生长情况。将细胞以适宜密度接种,加入含5-氮-2'脱氧胞苷的RPMI 1640培养基,使其药物最终浓度分别为0.5、2.5、12.5 μmol/L,每 24 h 更换新鲜药液,药物浓度同前,连续作用3 d后弃去药液,以含10%胎牛血清的RPMI 1640培养基继续培养5 d,以未加药物的细胞株作为对照组。
1.2 方法
1.2.1 MTT试验(四唑盐比色分析法) 上述经不同浓度药物处理3 d后的Reh细胞及对照组细胞,以每孔1×104接种于96孔培养板中,每孔体积100 μl,每组设3 个复孔,分别于第 24 h、48 h、72 h,于96 孔板中每孔加入 MTT(5 mg/ml,Sigma)20 μl孵育4 h。离心(1 000 rpm,5 min),弃上清。每孔加入二甲基亚砜(DMSO)150 μl,振荡 10 min,酶标仪490 nm处测吸光度A值。按下列公式计算细胞增殖抑制率:细胞增殖抑制率%=(1-实验组A值/对照组A值)×100%。
1.2.2 MSP检测RUNX3基因启动子区甲基化取上述经不同浓度药物处理3 d后的Reh细胞及对照组细胞1×106,每组设3个复孔,按DNA提取说明书(Promega公司)提取各组细胞基因组DNA,对提取的DNA用紫外分光光度计检测其纯度及含量,DNA 纯度测定:A260/A280>1.8。取2 μg DNA 按DNA甲基化试剂盒说明书进行修饰及纯化,纯化后的DNA立即使用或-20℃保存备用。纯化后的DNA作为模板进行MSP,甲基化特异性引物上游序列为 5'-TTACGAGGGGCGGTCGTACGCGGG-3',下游引物为 5'-AAAACGACCGACGCGAACGCCTCC-3',扩增片段为210 bp。非甲基化特异性引物上游序列为5'-TTATGAGGGGTGGTTGTATGTGGG-3',下游引物为 5'-AAAACAACCAACACAAACAACTCC-3',扩增片段为210 bp。PCR反应体系(25 μl):2×GoTaq®Hot Start Green Master Mix 12.5 μl,修饰后的 DNA模板 2 μl,双蒸水 8.5 μl,引物(10 μmol/L)各 1 μl。PCR反应条件:94℃预变性3 min后进行35个循环:94℃变性30 s,59℃退火30 s,72℃延伸30 s,最后一个循环后再72℃延伸5 min。每个样本扩增时,同时以灭菌双蒸水作为阴性对照。取5 μl PCR产物2%的琼脂糖凝胶(含溴化乙锭)电泳,分析产物。结果判断:仅出现甲基化条带为高甲基化,仅出现非甲基化条带为未甲基化,同时出现为半甲基化。
1.2.3 RT-PCR检测 RUNX3 mRNA的表达 取1×106细胞用Trizol试剂提取各组RNA,每组设3个复孔。用DNaseⅠ(RNA free)消化RNA中痕量的DNA,用紫外分光光度计及琼脂糖凝胶电泳检测RNA的含量及纯度。RNA的纯度测定:A260/A280>2.0。取消化后的RNA作为模板进行RT-PCR试验,操作按试剂盒说明书进行。RT-PCR体系为25 μl,2 × AccessQuickTMMaster Mix 12.5 μl,RNA 模板 2 μl,Nuclease-free water 6.5 μl,引物各 2 μl,AMV Reverse Transcriptase 0.5 μl。以 β-actin 作为内参照。RUNX3及β-actin内参照的引物序列如下:RUNX3上游序列为5'-TGGCAGGCAATGACG-3',下游序列为 5'CAGGGAACGGCTTGGT-3',产物为258 bp;β-actin上游序列为5'-CGCGAGAAGATACCCAGAT-3',下游序列为5'-GCACTGTGTTGGCGTACAGG-3',产物为550 bp。RT-PCR反应条件为:45℃孵育45 min,95℃预变性2 min之后进行35个如下循环:95℃变性30 s,52℃(RUNX3)60℃(β-actin)退火 30 s,72 ℃延伸 30 s,最后一个循环后再72℃延伸5 min。扩增产物2%的琼脂糖凝胶(含溴化乙锭)电泳,分析扩增产物。采用凝胶分析系统(捷达801系列)分析,以RUNX3/β-actin吸光度比值表示RUNX3 mRNA的相对水平。
1.2.4 SPSS13.0软件进行统计学处理与数据分析数值变量资料以±s表示,两组之间比较采用t检验或秩和检验;多组之间采用单因素方差分析(F检验)或秩和检验,P<0.05作为判断差异是否有统计学意义的依据。
2 结果
2.1 5-氮-2'脱氧胞苷对 Reh细胞增殖的影响MTT检测发现(表1),Reh细胞经3种不同浓度的5-氮-2'脱氧胞苷处理后,各组的细胞增殖抑制率如图1所示。可见,同一时间不同药物浓度之间的细胞增殖抑制率随浓度增大其抑制率增大,同一药物浓度不同时间组的细胞增殖抑制率随时间延长其抑制率也增大,两者均有统计学差异(P<0.05),5-氮-2'脱氧胞苷对Reh细胞增殖的抑制作用呈现时间和剂量的依赖性。
2.2 5-氮-2'脱氧胞苷处理前后RUNX3基因启动子甲基化状态改变 MSP检测Reh细胞用药前后RUNX3基因启动子区域甲基化状态,见图2。经0.5、2.5、12.5 μmol/L 不同浓度 5-氮-2'脱氧胞苷处理72 h后,对照组(未干预组)甲基化条带明显,而未甲基化条带隐约可见;在不同浓度5-氮-2'脱氧胞苷处理组,甲基化和未甲基化条带均明显可见,有甲基化和非甲基化共存现象,表明部分被去甲基化了。
表1 不同浓度5-氮-2'脱氧胞苷去甲基化干预在不同的作用时间对Reh细胞生长的抑制率(±s,n=3)Tab 1 The inhibitory rate of different concentrations and time on proliferation in Reh cells by demethylation agent 5-aza-2'-deoxycytidine
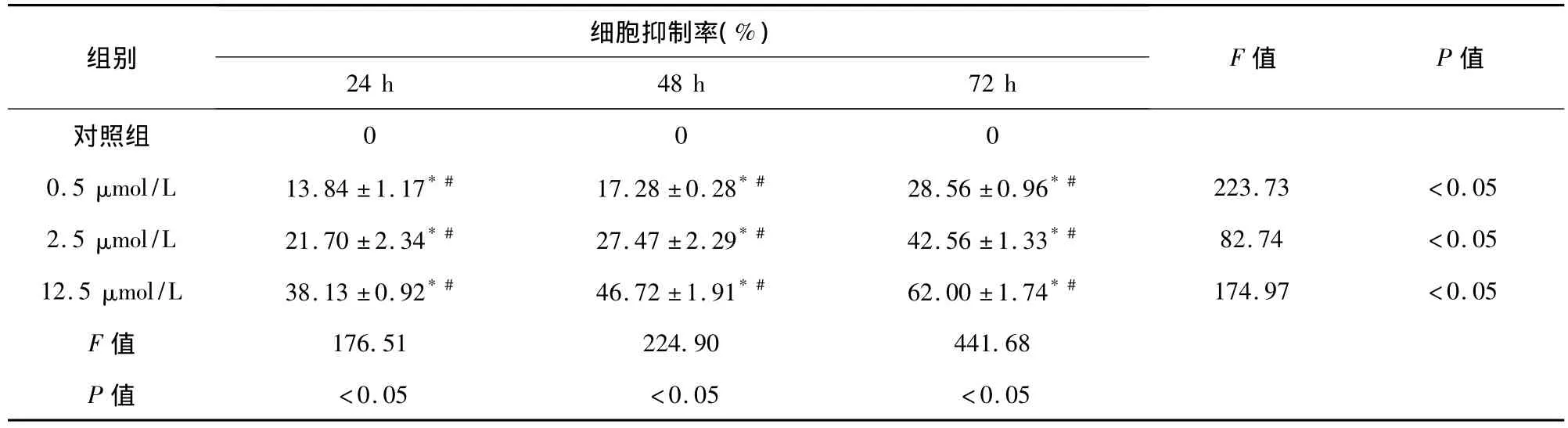
表1 不同浓度5-氮-2'脱氧胞苷去甲基化干预在不同的作用时间对Reh细胞生长的抑制率(±s,n=3)Tab 1 The inhibitory rate of different concentrations and time on proliferation in Reh cells by demethylation agent 5-aza-2'-deoxycytidine
注:*表示同一时间不同浓度之间两两比较有统计学意义,P<0.05;#表示同一浓度不同时间之间两两比较有统计学意义,P<0.05
组别 细胞抑制率(%)24 h 48 h 72 h F值 P值对照组000<0.05 <0.05 <0.05 0.5 μmol/L 13.84 ±1.17*# 17.28 ±0.28*# 28.56 ±0.96*# 223.73 <0.05 2.5 μmol/L 21.70 ±2.34*# 27.47 ±2.29*# 42.56 ±1.33*# 82.74 <0.05 12.5 μmol/L 38.13 ±0.92*# 46.72 ±1.91*# 62.00 ±1.74*# 174.97 <0.05 F 值 176.51 224.90 441.68 P值
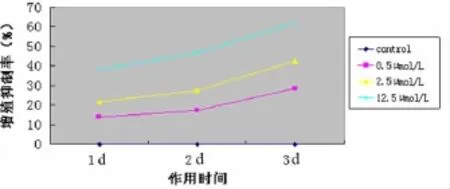
图1 不同浓度5-氮-2'脱氧胞苷去甲基化干预对Reh细胞生长的影响Fig 1 The inhibition of cell proliferation by demethylation agent 5-aza-2'-deoxycytidine of different concentrations in Reh cells
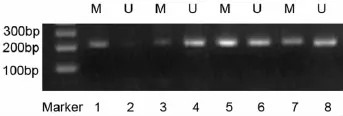
图2 Reh细胞经5-氮-2'脱氧胞苷干预72 h前后RUNX3基因启动区甲基化状态Fig 2 The methylation status of RUNX3 promoter region in Reh cells treated with 5-aza-2'-deoxycytidine before and after 72 hours
2.3 不同浓度5-氮-2'脱氧胞苷处理前后对RUNX3 mRNA表达影响 扩增产物经琼脂糖凝胶电泳显示,RUNX3 mRNA在对照组细胞中不表达,经0.5、2.5、12.5 μmol/L 不同浓度 5-氮-2'脱氧胞苷处理后可见RUNX3 mRNA重新表达,如图3所示,且其表达强度与5-氮-2'脱氧胞苷的浓度呈剂量依赖关系,相对表达量分别为,对照组:0.000±0.000;0.5μmol/L组:0.216 ±0.012;2.5 μmol/L 组:0.432±0.009;12.5 μmol/L 组:0.587 ± 0.020。各浓度组与对照组比较有统计学意义(P<0.05),各浓度组之间比较也有统计学意义(P<0.05)。

图3 不同浓度5-氮-2'脱氧胞苷干预72h前后RUNX3 mRNA的表达Fig 3 RUNX3 mRNA expression of Reh cells treated with 5-aza-2'-deoxycytidine before and after 72 hours
3 讨论
DNA甲基化作为表现遗传学的一种调控机制是最早被发现的[3],它在基因表达调控、细胞增殖、分化、发育及基因组印记等方面起着重要作用,并与肿瘤的发生发展关系密切[4]。急性白血病的发生是一个多步骤、多因素参与的复杂的生物学过程,越来越多的证据表明基因启动子区域CpG岛的高度甲基化也是白血病等血液系统恶性肿瘤的一种发病机制,大概有90%的血液系统恶性肿瘤都会有至少一个基因高度甲基化[5]。近年来许多研究表明,DNA甲基化状态异常尤其是抑癌基因的高甲基化失表达在白血病的发生发展、诊断、检测微小残留病、预测病情及指导治疗方面都有重要作用[6~8]。本研究室近年也在白血病基因治疗方面做了大量工作[9,10]。
RUNX3基因是Levanon等于1994年发现的抑癌基因,研究发现,RUNX3蛋白可指导TCF-β信号转导过程中激活的Smad复合物从细胞质内转入细胞核内特定靶位点,加强Smad复合物与靶位点的结合强度并激活靶基因,从而对细胞的分化、细胞周期调控、凋亡和恶性转化起作用[11]。
本研究选用5-氮-2'脱氧胞苷作为甲基化制剂处理白血病Reh细胞株,5-氮-2'脱氧胞苷可通过抑制DNA甲基转移酶1(DNMT1)的甲基转移活性从而达到去甲基化,使多种含有CpG岛的高甲基化抑癌基因重新表达或增强,从而恢复抑癌功能,如P16基因[12]。本研究结果显示,MTT法检测到经过不同浓度5-氮-2'脱氧胞苷处理后,Reh细胞生长增殖活性受到了抑制,且细胞增殖抑制率随着药物浓度增大和作用时间延长而增强。MSP法检测到RUNX3基因启动子在5-氮-2'脱氧胞苷未干预的Reh细胞中是甲基化的,RT-PCR法检测到 Reh细胞中RUNX3 mRNA未见表达,经过不同浓度5-氮-2'脱氧胞苷处理后,RUNX3基因启动子甲基化区域出现部分去甲基化,而RUNX3 mRNA可见重新表达。当然5-氮-2'脱氧胞苷本身存在的细胞毒性也可能导致类似作用,目前研究表明,有细胞毒性的药物,其细胞毒性均存在时效性,而未发现抑制肿瘤细胞生长的可遗传性[13]。本实验所用Reh细胞是5-氮-2'脱氧胞苷作用后继续培养5 d以上的,证实Reh细胞生长增殖活性的抑制作用并非由5-氮-2'脱氧胞苷本身药物毒性直接决定的,而是去甲基化的间接作用,从而进一步论证了抑癌基因RUNX3不表达的主要机制是RUNX3基因启动子的异常甲基化,RUNX3基因启动子区异常甲基化参与了急性白血病的发生发展过程,去甲基化抑制剂5-氮-2'脱氧胞苷通过重新激活因甲基化处于沉默的RUNX3抑癌基因的转录活性,恢复了RUNX3基因的表达,因此RUNX3启动子的异常甲基化可能是急性白血病发病机制之一。
本实验结果表明去甲基化药物5-氮-2'脱氧胞苷能够使白血病Reh细胞RUNX3基因甲基化程度降低,激活RUNX3基因重新表达,抑制白血病细胞的生长增殖,为RUNX3成为白血病基因治疗的新靶点提供分子依据,从而为进一步用诱导沉默的RUNX3基因重新表达的方法对白血病进行基因治疗打下理论基础。
[1]林福安,叶宝国,沈建箴,等.半巢式甲基化特异性聚合酶链反应对肿瘤细胞株p15基因甲基化或缺失状态的检测[J].中国实验血液学杂志,2007,15(2):382 -386.
[2]LI QL,ITO K,SAKAKURA C,et al.Causal Relaationship between the loss of Runx3 Expression and Gastric Cancer[J].Cell,2002,109(1):113-124.
[3]LI E.Chromatin modification and epigenetic reprogramming in mammalian development[J].Nat Rev Genet,2003,3:662 - 673.
[4]EHRLICH M.The controversial denouement of vertebrate DNA methylation research[J].Biochemistry,2005,70:568 -575.
[5]陈丹青.DNA甲基化与白血病[J].医学理论与实践,2011,24(6):635-636.
[6]DAS PM,SINGAL R.DNA methylation and cancer[J].J Clin Oncol,2004,22(22):4632 -4642.
[7]史晓红,范芸,周昌虎,等.急性髓系白血病SFPR4基因启动子区异常甲基化研究[J].中华临床医师杂志,2011,5(4):990-995.
[8]VALENCIA A,ROMAN GOMEZ J,CERVERA J,et al.Wnt signaling pathway is epigenetically regulat-ed by methylation of Wnt antagonists in acute myeloid leukemia[J].leukemia,2009,23(9):1658-1666.
[9]吴明彩,蒋明,毕富勇.脂质体转染STAT3反义核酸对HL-60细胞增殖和凋亡的影响[J].皖南医学院学报,2008,27(6):448-451.
[10]吴明彩,吕俊,戚之琳,等.HHT联合低剂量Ara-c与RNA诱导K562细胞凋亡的研究[J].皖南医学院学报,2007,26(1):15-17.
[11]ZAIDI SK,SULLIVAN AJ,VAN WIJNEN AJ,et al.Integration of Runx and Smad regulatory signals at transcriptionally active subnuclear sites[J].Proc Natl Acad Sci USA,2002,99(12):8048 -8053.
[12]KANTARJIAN H,OKI Y,GARCIA-MANERO G,et al.Results of a randomized study of 3 schedules of low-dose decitabine in higherrisk myelodysplastic syndrome and chronic myelomonocytic leukemia[J].Blood,2007,109(1):52 - 57.
[13]BENDER CM,PAO MM,JONES PA.Inhibition of DNA methylation by 5-aza-2'-deoxycytidine suppresses the growth of human tumor cell lines[J].Cancer Res,1998,58(1):95 - 101.
猜你喜欢
现代农村科技(2022年1期)2022-01-21
中国循证心血管医学杂志(2021年10期)2021-11-05
台州学院学报(2018年6期)2018-02-26
河南畜牧兽医(2017年12期)2017-11-13
中国科技信息(2015年6期)2015-11-10
癌变·畸变·突变(2015年3期)2015-02-27
现代检验医学杂志(2015年2期)2015-02-06
高中生学习·高三版(2014年3期)2014-04-29
遗传(2014年3期)2014-02-28
中国海洋大学学报(自然科学版)(2014年12期)2014-02-28
