
复方绞股蓝胶囊质量标准研究
2011-05-31 02:51熊野娟
中国药业 2011年15期
熊野娟
(上海医药高等专科学校药学系,上海 201318)
复方绞股蓝胶囊含有绞股蓝总苷和灯盏花素,具有提高机体免疫能力、活血化瘀的功效[1],主要用于治疗口腔黏膜白斑,而口腔黏膜白斑是国际公认最常见的口腔黏膜癌前病变。原质量标准中用紫外分光光度法进行含量测定,为提高制剂质量的可控性,笔者采用薄层色谱法对绞股蓝进行定性鉴别,采用高效液相色谱法对灯盏花素中主要成分野黄芩苷进行含量测定,现将结果报道如下。
1 仪器与试药
Agilent 1100型高效液相色谱仪,DAD检测器;AB 204-N型电子天平、AG-135型电子天平(梅特勒上海有限公司)。野黄芩苷对照品(中国药品生物制品检定所,批号为110842-200605);绞股蓝皂苷(西安小草植物科技有限责任公司,批号为XC100301);复方绞股蓝胶囊(上海第九人民医院);硅胶G(青岛海洋化工厂);乙腈、甲醇和磷酸为色谱纯,水为超纯水,其他试剂均为分析纯。
2 方法与结果
2.1 绞股蓝定性鉴别(薄层色谱法)
称取复方绞股蓝胶囊内容物1 g,加三氯甲烷40 mL,加热回流5 h,弃去滤液,药渣挥干溶剂,加水0.5 mL,搅匀湿润后,加水饱和的正丁醇10 mL,超声处理30 min,吸取上清液,加3倍量的氨试液,摇匀,放置分层,取上层液蒸干,残渣加甲醇10 mL使溶解,作为供试品溶液。取不含灯盏花素的阴性样品,同法制备阴性对照品溶液。精密称取绞股蓝皂苷对照品2 mg,置1 mL容量瓶中,加甲醇使溶解并稀释至刻度,作为对照品溶液。照2010年版《中国药典(一部)》附录ⅥB薄层色谱法试验,吸取上述溶液各10 μL,分别点于同一硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇-水(15∶40∶20∶10)10℃以下放置分层的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,105℃加热至斑点显色清晰。结果供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的斑点,阴性对照品溶液无干扰(图1)。

图1 薄层色谱图
2.2 野黄芩苷含量测定(高效液相色谱法)
2.2.1 色谱条件与系统适用性试验
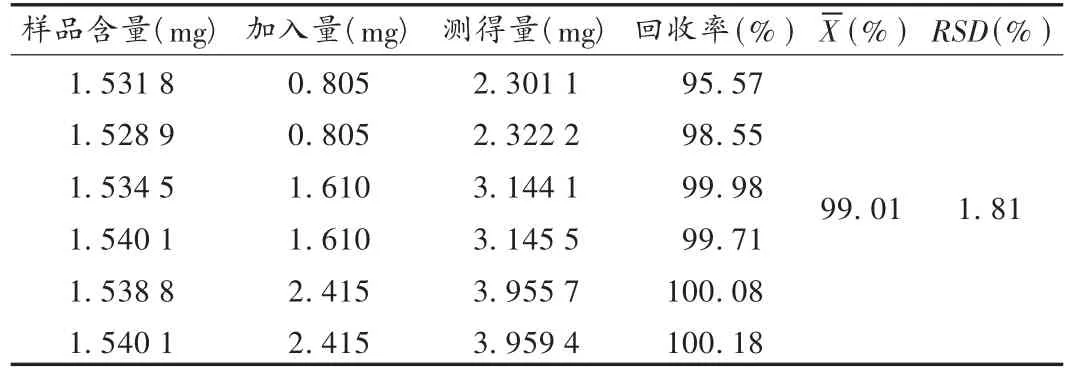
表1 野黄芩苷加样回收试验结果(n=6)

图1 高效液相色谱图
色谱柱∶Kromasil 100-5 C18柱(150 mm×4.5 mm,5 μm);流动相∶乙腈 -0.1%磷酸(15 ∶85);检测波长∶335 nm;柱温∶35℃;流速∶1.0 mL/min;进样量∶10 μL。在此条件下,野黄芩苷与其他组分峰可达基线分离,且与相邻色谱峰分离度大于1.5,理论板数按野黄芩苷计算均在3 000以上。
2.2.2 溶液制备
精密称取野黄芩苷对照品9.005 0 mg,置50 mL容量瓶中,加甲醇使溶解并稀释至刻度,制成每1 mL含野黄芩苷0.180 1 mg的对照品贮备液。精密量取贮备液10 mL,置25 mL容量瓶中,加甲醇稀释至刻度,摇匀,得野黄芩苷对照品溶液。精密称取复方绞股蓝胶囊内容物40 mg,置50 mL容量瓶中,加甲醇适量,超声处理15 min,放冷,加甲醇定容至刻度,摇匀,滤过,取续滤液作为供试品溶液。取缺野黄芩苷的其余处方量药材,按所确定的制备工艺制备缺野黄芩苷的阴性样品,按供试品溶液制备方法制备缺野黄芩苷的阴性对照品溶液。
2.2.3 方法学考察
阴性干扰试验∶分别精密吸取对照品溶液、阴性对照品溶液与供试品溶液各10 μL,注入液相色谱仪,测定。结果表明,色谱行为良好,各色谱峰均达基线分离;供试品溶液色谱中,在与对照品溶液色谱峰相应的位置上检出色谱峰,而缺野黄芩苷的阴性对照品溶液色谱中无相应色谱峰,表明阴性对照品无干扰(图1)。
线性关系考察∶分别精密量取对照品贮备液 1,5,10,15,20 mL,置25 mL容量瓶中,加甲醇稀释至刻度,摇匀,按2.2.1项下的条件测定,以峰面积为纵坐标、对照品质量浓度为横坐标进行线性回归,得回归方程 Y=28 870.944 15X -16.489 20,r=0.999 94(n=6)。结果表明,野黄芩苷质量浓度在0.007~0.180 0 g/L范围内与峰面积线性关系良好。
精密度试验∶取同一质量浓度(0.180 1 g/L)的野黄芩苷对照品溶液10 μL,连续进样6次,按上述色谱条件测定,野黄芩苷峰面积的 RSD=0.09%(n=6),表明仪器精密度良好。
稳定性试验∶取同一供试品溶液,在 0,2,4,6,8,10,12 h 时分别进样,结果峰面积积分值平均为2 059.577 56,RSD=1.4%(n=7),表明供试品溶液在12 h内稳定。
重复性试验∶取同一批号的样品5份,依法制备供试品溶液并按上述色谱条件测定。结果平均峰面积为1 905.592 36,RSD=1.5%(n=5),表明方法重复性良好。
加样回收试验∶精密称取对照品10.062 5 mg,置25 mL容量瓶中,加甲醇使溶解并稀释至刻度,摇匀,再精密吸取20 mL,置50 mL容量瓶中,加甲醇稀释至刻度,制成每1 mL含0.161 0 mg的溶液。称取已知含量的6份样品,精密称定,置50 mL容量瓶中,分别加入上述对照品溶液5,10,15 mL,按样品测定法测定含量,结果见表1。
2.2.4 样品含量测定
取复方绞股蓝胶囊样品适量,照2.2项下供试品溶液制备方法制备溶液,按上述色谱条件进行含量测定。结果批号为20080628,20080717,20080901的3批样品中,野黄芩苷的含量分别为7.13,7.23,7.18 mg/g,平均 7.18 mg/g,RSD 为 1.35%(n=2)。
3 讨论
对绞股蓝进行薄层色谱鉴别时,比较了三氯甲烷-乙酸乙酯-甲醇(20∶45∶35)、三氯甲烷 -乙酸乙酯 -甲醇 -水(3∶8∶4∶2)[2]、三氯甲烷-乙酸乙酯-甲醇 -水(15∶40∶20∶10)3种展开剂,结果只有最后1种展开剂的下层液能使绞股蓝皂苷色谱斑点与相邻色谱斑点分离,且斑点清晰。
在对野黄芩苷的含量进行测定时,考察了流动相和柱温的条件,试用乙腈 - 水(35 ∶65)[3]、乙腈 -0.1% 磷酸(25 ∶75)、乙腈 -0.1%磷酸(15∶85)3种流动相,发现只有乙腈-0.1%磷酸(15∶85)为流动相且在柱温为35℃时,野黄芩苷峰与相邻色谱峰达到有效分离,且色谱行为良好。方法学考察结果表明,高效液相色谱法重复性好,精密度和回收率都较满意,因此可作为复方绞股蓝胶囊的质量控制方法。
[1]李群峰.绞股蓝的化学成分和药理作用研究进展[J].光明中医,2009,24(12):2 396 -2 397.
[2]王瑞雪,王树华,王建辉,等.长梗绞股蓝皂苷B的分离[J].华北煤炭医学院学报,2008,10(3):340-341.
[3]张转平,王新权,胥道宝,等.HPLC法测定绞股蓝中的绞股蓝皂苷A的含量[J].西北药学杂志,2008,23(2):87 -88.
猜你喜欢
中国应急管理科学(2021年4期)2021-04-13
中学生数理化·高一版(2020年9期)2020-01-02
中学化学(2019年4期)2019-08-06
——一个解释欧姆表刻度不均匀的好方法
教学考试(高考物理)(2018年6期)2018-12-06
考试周刊(2018年68期)2018-09-17
中成药(2018年1期)2018-02-02
恋爱婚姻家庭·养生版(2017年12期)2017-12-07
恋爱婚姻家庭(2017年36期)2017-07-22
学苑创造·B版(2017年1期)2017-02-21
学苑创造·B版(2017年1期)2017-02-21
