
一个遗传性多巴反应性肌张力障碍家系的临床特点及GCH-Ⅰ基因突变分析☆
2011-02-01 03:32薛原陈先文高宗良严孙宏
中国神经精神疾病杂志 2011年4期
薛原 陈先文 高宗良 严孙宏
一个遗传性多巴反应性肌张力障碍家系的临床特点及GCH-Ⅰ基因突变分析☆
薛原*陈先文*高宗良*严孙宏*
目的 探讨一遗传性多巴反应性肌张力障碍(dopa-responsive dystonia,DRD)家系临床特点和相关基因突变。方法 收集一个多巴反应性肌张力障碍家系全部家庭成员临床资料并随访,采集家系成员静脉血,常规提取基因组DNA,利用PCR技术扩增三磷酸鸟苷环化水解酶Ⅰ(guanosine triphosphate cyclohydrolase 1,GCH-Ⅰ)基因全部6个外显子,进行DNA直接测序并分析。结果 该家系患病成员临床主要表现为肌张力障碍和帕金森综合征,但症状严重程度个体差异大,所以患者对多巴丝肼治疗均有较好反应,1例患者长期大剂量不规律使用左旋多巴后出现异动症。基因测序显示4例患病成员GCH-Ⅰ第四外显子102号碱基由胸腺嘧啶替换为腺嘌呤(T→A),并导致176位丝氨酸(Ser)转变为精氨酸(Arg)Arg176 Ser,该突变类型尚未见文献报道。其余家系成员未发现GCH-Ⅰ基因突变。结论 多巴反应性肌张力障碍临床表现在同一家系不同个体之间可有较大差异,部分患者长期大量不规律使用左旋多巴亦可导致异动症产生。GCH-Ⅰ基因第四外显子102号碱基(T→A)突变是DRD患病的分子遗传学原因之一。
多巴反应性肌张力障碍 临床表现 GCH-Ⅰ基因 突变
多巴反应性肌张力障碍(dopa-responsive dystonia,DRD)是一种临床较为罕见的遗传性神经疾病,可以是常染色体显性遗传,也可表现为常染色体隐形遗传。其临床表现有两个显著区别于其他肌张力障碍的特点,即DRD患者肌张力变化呈昼夜波动,同时小剂量多巴胺治疗可使症状获得戏剧性改善并可长期维持,多巴胺不良反应罕见[1]。该病发病范围较广,全球均有报道。已知90%突变集中在 GCH-Ⅰ(guanosine triphosphate cyclohtdrolase 1,GCH-Ⅰ三磷酸鸟苷环化水解酶Ⅰ)基因[1-3]。在我国,发生在 GCH-Ⅰ基因外显子1,3,6中的突变均有报道,分别为发生在第一外显子非编码区的 T71C及 G151A,,外显子 3中的Gln161Pro及外显子6的Lys224Arg。同时,发生在内含子的基因突变亦有1例为IVS5+3insT[4-7]。本文对一个临床诊断的DRD家系的临床表现和GCH-I基因进行分析,以助了解中国人家族性DRD临床特征和相关的分子遗传学特征。
1 资料与方法
1.1 家系资料 研究对象为安徽省庐江县一DRD患病家系,临床诊断参照文献标准[8]。该家系见图1。现存家庭成员12名,第一代父亲已亡,具体原因不详,母亲为健康个体,第二代家庭成员现存6名,患病者4例,男1例,女3例,病程长者已达37年,短者12年,健康个体2名,男女各有1名。另有两名第二代家庭成员已亡故,均系女性,具体原因不详。第三代现存成员5名,女1名,男4名,尚无发病个体。家族祖父母,及其他具有血亲关系个体未见具有类似症状者。
1.2 基因分析方法 获得知情同意后,收集本家系母亲及现存所有二代成员外周静脉血各2 mL,EDTA抗凝。-80℃保存备用。使用北京索来宝生物工程有限公司哺乳动物全血DNA提取试剂盒(吸附柱法)常规分离白细胞后提取基因组DNA,作为PCR反应模板分装 保 存。根 据 http://www.ncbi.nlm.nih.gov/pubmed.数据库查取人GCH-I基因DNA序列,扩增全部6个外显子,引物设计参照Bandmann等人设计的引物[9].引物序列,及扩增片段如下表。引物由上海生工生物工程有限公司合成。PCR反应体系配置为50 μL,其中基因组 DNA 1 μL(200 ng/μL),上下游引物各 2 μL(30 ng/μL),MgCl25 μL(25 mmol/L),dNTP 10μL(25 mmol/L),10×PCR buffer 10μL(100 mM Tris-HCl,500 mM KCl),Taq DNA Polymerase 5 μL(1 umol/L),ddH2O 25μL。PCR反应条件:94℃预变性5 min,随后以94℃变性30 s,退火45 s(不同引物退火温度不同),72℃延伸45 s,反应35个循环后72℃延伸10min。PCR反应产物以4℃保存,经1%琼脂糖电泳检测扩增成功,PCR产物经纯化后直接进行DNA测序,全部测序由上海生工测序部完成。
2 结果
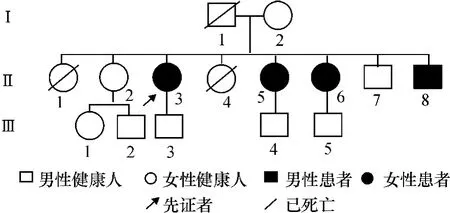
图1 该DRD家系的遗传图谱

表1 PCR反应引物序列及扩增片段长度
2.1 临床特点 病例1(图1中Ⅱ3):先证者,女,现年45岁,7岁起病,以步态不稳,易摔倒为首发症状,左侧肢体较明显,症状至下午加重,睡眠后可获部分改善。发病后十年间逐渐出现双下肢僵硬,活动不灵活,行走不稳,易摔倒。并且症状逐渐波及双上肢,下午至夜间症状明显加重,同时伴有咬字不清,进食缓慢,饮水呛咳。30余岁后上述症状基本稳定,晨轻暮重现象已不明显。查体:身材较矮小,反应迟钝,构音不清,咽反射减弱。四肢肌张力齿轮样增高,动作迟缓,内翻足,双足趾跖屈,踝关节和足趾关节有挛缩变形,四肢腱反射活跃,病理反射未引出。3年前开始使用多巴丝肼治疗,125 mg每天3次,服药1周后,上述肢体僵硬症状明显改善。1个月后,构音明显改善,进食顺利,四肢自主活动能力明显增强,肌张力略低,生活可基本自理。追踪回访,该患者于2年后出现四肢肌张力低,并伴有舞蹈样粗大不自主动作。详细询其问服药及生活情况,病人自述为使症状继续好转,自行加大多巴丝肼剂量至250 mg每天3次,并有同时加服安坦等其他药物情况,且服药不规律,时服时断,4年后出现双上肢不自主运动,自己不能控制,睡眠后可消失。给予患者减少多巴丝肼用量至原剂量,停用其他药物,并嘱患者勿自行增减药物,舞蹈症状消失。随访至今未再出现异动症,目前多巴丝肼维持剂量187.5 mg每天3次,安坦2 mg每天3次,生活部分自理,可持仗行走。
病例2(图1中Ⅱ5):女,42岁,36岁起病,主要表现为双下肢僵硬不适感,左侧明显,行走稍欠灵活,其余无明显异常,口服多巴丝肼125 mg每日1次,1周后双下肢僵硬明显改善,步态正常。目前持续治疗已3年,未见不良反应。
病例3(图1中Ⅱ6):女,40岁,13岁起病,起病状态与病例1类似,查体见:表情呆滞,反应略迟钝,肢体肌张力铅管样增高,肢体轮替动作迟缓笨拙,腱反射弱,病理反射未引出。四肢关节轻度挛缩变形,肌肉废用性萎缩。治疗给予口服多巴丝肼125 mg每日3次,1个月后,患者活动能力明显改善,可独立缓慢行走,生活基本自理。无不良反应出现。
病例4(图1中Ⅱ8):男,33岁,12岁左右发病,同样以双下肢活动不自如起病,逐渐进展。现四肢有僵硬感,双上肢动作笨拙。行走步态不稳,起坐需扶物。查体:四肢肌张力铅管样增高,肢体轮替动作迟缓,腱反射正常,病理征未引出。予口服多巴丝肼125 mg每天3次,1周后四肢肌张力基本正常,仅存步态轻度不稳。至今症状平稳无其他不适症状。
本家系已知4例患病成员均来自同一代,男女均有发病,其母表型正常,其父早年病故,死因不详。患病成员均慢性起病,在数年内症状逐渐加重,主要表现为下肢肌张力障碍和帕金森综合征症状,但进展速度和症状严重程度个体差异大。病例1(Ⅱ3)发病年龄早(7岁),症状最重,除肌张力障碍症状外,还有突出的帕金森综合征表现,由于家境贫困未及时诊治,至诊断时已部分致残,生活不能自理,治疗后仍遗留部分功能残疾。病例4(Ⅱ8)和病例3(Ⅱ6)临床表现与病例1类似,但发病年龄较晚,症状较轻。病例2(Ⅱ5)36岁才开始发病,症状最轻,仅有轻度下肢肌张力障碍,无明显帕金森综合征症状。
本家系所有患者对左旋多巴治疗均有较好反应,但2名患者由于未得到及时诊治,已出现关节挛缩变形和肌萎缩,严重影响运动功能,在药物治疗前日常生活无法自理,经多巴丝肼加安坦治疗后,虽然症状得到显著改善,但仍遗留部分肢体功能残疾,日常生活能力也没有恢复到正常水平。其余2例药物治疗后功能基本恢复正常水平。本家系病例1(Ⅱ3)由于自行不规则使用大剂量多巴丝肼出现较严重的异动症症状,药物减量后异动症症状消失。
2.2 GCH-I基因分析 在该家系,第一代母亲GCH-Ⅰ基因外显子部分经测序未发现异常,第二代4例患病者均发现在第四个外显子第102号碱基由T转换为A,使176位丝氨酸(Ser)转变为精氨酸(Arg)(图2)。其余未患病家庭成员均未检测出相同突变。
3 讨论
本家系同一代中出现4例患病者,女3例,男1例,同代其他堂表兄妹中亦无患病个体。上一代家庭成员中母亲表型正常且GCH-I基因分析未发现异常,其父病故,死因不详,遗传模式考虑为常染色体显性遗传,突变基因可能遗传自其父亲。初始症状出现年龄从7岁到33岁,相差较大。临床表现主要为肌张力障碍及帕金森综合征,累及四肢躯干以及咽喉肌,起病初期具有晨轻暮重现象。个体间症状表现类似但程度明显不同,病例1(Ⅱ3)7岁发病,症状最重,病例3(Ⅱ6)和病例4(Ⅱ8)在13~14岁发病,症状中等,病例2(Ⅱ5)36岁才开始发病,症状最轻,提示本病症状轻重与发病年龄有关,发病越早,症状越重,儿童期患病病例身体发育状况明显差于成年期患病个体。上述临床特点均与国内外报道一致。
多巴反应性肌张力障碍虽然为一种遗传性疾病,但积极治疗可获得较为满意的疗效,使患者生活质量明显提高。在国内外文献报道中,左旋多巴是治疗该病的首选,小剂量即长期有效且无运动并发症,本家系所有患者对左旋多巴治疗有较好反应,但有2例患者未能及时得到诊治,导致关节不同程度的挛缩变形,虽经治疗症状得到明显改善,但仍遗留有功能残基,1名症状较重的患者需要使用较大剂量多巴丝肼症状才能得到较好控制。这一结果说明对DRD患者早期诊断、及时治疗的重要性。异动症为左旋多巴治疗帕金森病时常见的不良反应,但在已有报道的DRD患者中出现极为罕见,值得注意的是该家系1名患者(病例1)在长期大剂量、不规律多巴丝肼治疗后出现了肢体异常不自主运动症状,在药物减量后症状消失,提示DRD患者如果药物治疗不当亦可引起左旋多巴诱发的异动症,值得临床工作中注意。
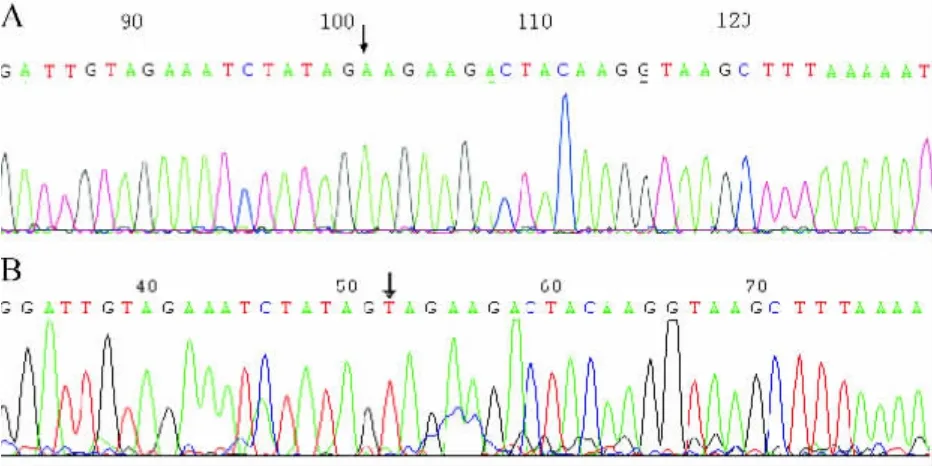
图2 GCH-I基因外显子4点突变测序图。A:患病成员GCH-I基因外显子4点突变测序图;B:未患病成员GCH-I基因外显子4正常序列对照
1994年,Nishivama等应用基因连锁方法将该疾病的致病基因定位于第14号染色体上的三磷酸鸟苷环化水解酶1,(GTP cyclohydrolase 1 GCH-I)的 Iq22.1 -22.2区域。目前世界范围内报告引起DRD发病GCH-Ⅰ基因突变有100余种,绝大部分集中在GCH-I基因的六个编码蛋白质的外显子区域,部分突变发生在内含子及结合部(http://www.hgmd.org/)。同时,根据已有研究,导致DRD发病的致病基因还有TH基因,通常表现为常染色体隐性遗传方式,其临床表现与GCHI基因突变者有一定差别。此外,仍有30%~40%的患者未能在在上述两种基因中检测到任何形式的基因突变。本家系基因测序显示母亲为正常个体,子代四例患者均可见到GCH-I基因第四个外显子(exon4)上第102号碱基发生T→A的替换,引起蛋白序列第176位中性氨基酸丝氨酸(Ser)转变为碱性氨基酸精氨酸(Arg),引起GCH-I酶结构和功能异常,活性下降。国外文献有相同位点突变报道,但突变形式不同,为Thr176Ser(G→C)[11],国内尚未出现相同位点突变报道。该患病病例为常染色体显性异常的子代杂合子。Ichionose等[12]对已知GCH-I基因异常的杂合子可引起GCH-I酶活性进行研究,提示携带该基因异常杂合子的个体体内GCH-I酶活性下降均在50%以上,如携带者酶活性下至20%左右甚至更低,则引起DRD临床症状,部分携带者酶活性可维持在正常值的30%~40%,成为无症状携带者。
[1] Nygaard TG,Wilhelmse KC,Risch NG,etal.Linkagemapping of dopa-responsive dystonia(DRD)to chromosome 14q[J].Nat Genet,1993,5(4):386 -391.
[2] Nygaard TG,Marsden CD,Duvoisin RC.Dopa-responsive dystonia[J].Adv Neurol,1988,50:377 -384.
[3] Segawa M.Hereditary progressive dystonia with marked diurnal fluctuation[J].Brain Dev,2000,22(S):S65 - S80.
[4] 张社卿,郑惠民,谢惠君,等.国人多巴反应性肌张力障碍的分子遗传学研究[J].第二军医大学学报,2002,23(11):1233-1236.
[5] 周海燕,张本恕.鸟苷酸环化水解酶Ⅰ基因突变与相关表现型[J].国外医学分子生物学分册,2003,25(4):248-250.
[6] 刘燕玲,宋小华,李国林.多巴反应性肌张力不全临床研究[J].实用儿科临床杂志,2003,18(11):909-910.
[7] 李静,唐北沙,郭纪锋,等.多巴反应性肌张力障碍GCH1基因突变分析[J].中华医学遗传学杂志,2007,24(3)192-194.
[8] 梁秀龄.多巴反应性肌张力障碍[M]∥刘焯霖,梁秀龄,张成,主编.神经遗传病学.第2版.北京:人民卫生出版社,2002:180-182.
[9] Bandmann O,Nygaard TG,Surtees R,et al.Dopa-responsive dystonia in British patients:new mutations of GTP-cyclohydrolase I gene and evidence for genetic heterogeneity[J].Hum Mol Genet,1996,5(3):403-406.
[10] Fkawa Y,Graf WD,Wong H,et al.Dopa-responsive dystonia simulating spastic paraplegia due to tyrosine hydroxylase(TH)genemutations[J].Neurology,2001,56(2):260 - 263.
[11]Ilarioshkin SN,Markova ED,Slominsky PA,etal.The GTP cyclohydrolase Igene in Russian familieswith dopa-responsive dystonia[J].Arch Neurol,1998,55(6):789 -792.
[12]Ichionose H,Inagakii N,Suzuki S,et al.Molecularmechanisms of Hereditary Progressive Dystoniawithmarked diurnal fluctuation,Segawa's Disease[J].Brain Dev,2000,22(S):107 -110.
The clinical features and identification of a novel GCH-1 genemutation in a Chinese fam ily with dopa-respon- sive dystonia.
XUE Yuan,CHEN Xianwen,GAO Zongliang,YAN Sunhong.Department of Neurology,The First Affiliated Hospital of AnhuiMedical University.218 Jixi Road,Hefei.230022.China.Tel:0551 -2922114.
ObjectiveTo study the clinical features of dopa-responsive dystonia(DRD)and guanosine triphosphate cyclohydrolase 1(GCH-1)genemutation in a Chinese family.Methods This family had been followed up for three years after confirmation of diagnosis.Blood samples were collected from the familymembers and their genome DNA was extracted by standard technique.PCR and DNA sequencer were used to amplify all of6 exons of GCH-1 gene,and sequence PCR products,respectively.Resu lts The clinical features weremainly dystonia and parkinsonism.But the symptom severity varied among affected individuals.All affected familymembers had quite good response to levedopa treatment except one patient developed severe dyskinesia after a long-term high-dose treatment,which was rarely reported before.DNA sequencing showed a novel pointmutation with transition of T to A at the 102 base of exon 4 of GCH-1 gene,resulting in a substitution of arginine by serine at pointof176 in the GCH-1 protein in the 4 affectedmembers.Conclusions The symptom severity of DRDmay vary significantly among individuals even in the same family.In rare cases,patient can developed severe dyskinesia after long-term improper high-dose levedopa therapy.Mutation wit T→A at point102 of exon 4 in GCH-1 gene should be considered as a novel pathogenic mutation for dopa-responsive dystonia.
Dopa-responsive dystonia Clinical features Guanosine triphosphate cyclohydrolase 1(GCH-1)gene Mutation
R596
A
☆安徽省科技厅科技项目(编号:10021303036)
* 安徽医科大学第一附属医院神经内科(合肥 230022)
E-mail:chxwmail@yahoo.com.cn)
2010-10-12)
(责任编辑:李 立)
·短著述·
猜你喜欢
全科护理(2022年3期)2022-02-18
中国康复(2021年6期)2021-11-30
世界最新医学信息文摘(2021年12期)2021-06-09
中国保健营养(2019年7期)2019-10-21
民族古籍研究(2018年1期)2018-05-21
中国继续医学教育(2018年9期)2018-04-03
中西医结合心血管病杂志(电子版)(2017年31期)2018-01-04
中国现代神经疾病杂志(2017年1期)2017-03-29
中国神经免疫学和神经病学杂志(2014年5期)2014-05-08
中医研究(2014年6期)2014-03-11
