
蒽类衍生物的电荷传输性质
2010-12-05 02:27段桂花高洪泽王丽娟张厚玉马於光
物理化学学报 2010年8期
段桂花 高洪泽 王丽娟 张厚玉,* 马於光,*
(1吉林大学超分子结构与材料国家重点实验室,长春 130012;2中国人民武装警察部队学院基础部,河北廊坊 065000)
蒽类衍生物的电荷传输性质
段桂花1高洪泽2王丽娟1张厚玉1,*马於光1,*
(1吉林大学超分子结构与材料国家重点实验室,长春 130012;2中国人民武装警察部队学院基础部,河北廊坊 065000)
以具有较高迁移率的对称取代类蒽的衍生物{2,6-二[2-(4-戊基苯基)乙烯基]蒽,DPPVAnt;2,6-二-噻吩蒽,DTAnt;2,6-二[2-己基噻吩]蒽,DHTAnt}为研究对象,采用密度泛函理论的B3LYP方法,在6-31G(d)的基组水平上研究了三种蒽类衍生物的分子结构、电子结构、重组能和电荷传输积分,采用Einstein关系式计算了室温下的载流子迁移率,并与蒽的相关计算结果进行了比较.DPPVAnt是较好的空穴传输材料,其空穴迁移率为0.49 cm2·V-1·s-1;DHTAnt有利于电子传输,其电子迁移率为0.12 cm2·V-1·s-1;而DTAnt是一种较好的双极性材料,其空穴迁移率和电子迁移率分别为0.069和0.060 cm2·V-1·s-1.计算得到的迁移率与实验结果处于同一数量级.三种蒽类衍生物的电子重组能与蒽的相近,而空穴重组能均大于蒽的空穴重组能,大小顺序为蒽<DPPVAnt<DTAnt<DHTAnt.这与计算的迁移率结果不一致,说明分子的堆积结构决定材料的电荷传输性质.
密度泛函理论;蒽类衍生物;迁移率;电荷传输;分子重组能
近年来,随着有机半导体材料在有机电致发光二极管、薄膜晶体管、太阳能电池和激光器等领域研发的不断深入,人们逐渐认识到影响有机半导体电子器件性能的一个重要参数是载流子的迁移率[1-5].迄今为止,在薄膜晶体管中应用最为广泛的是并五苯及其衍生物,其场效应迁移率高达3.0 cm2·V-1·s-1[6].但是,以并五苯为基础的有机薄膜晶体管材料的空气稳定性非常有限,影响器件的寿命.利用蒽具有较宽带隙的特点来提高材料的稳定性,Meng等[7-8]合成了三种对称取代的蒽的衍生物{2,6-二[2-(4-戊基苯基)乙烯基]蒽,DPPVAnt;2,6-二-噻吩蒽,DTAnt; 2,6-二[2-己基噻吩]蒽,DHTAnt},其优化后的结构示意图如图1所示),且这些衍生物具有较高的载流子迁移率.载流子迁移率在很大程度上依赖于实验测定的条件和方法,而理论模拟可以研究有机半导体材料中电荷输运机制和计算材料的本征载流子迁移率,能够从更微观的角度去理解、印证和预测实验的测量数据.Deng等[9]采用Marcus-Hush两态模型的方法,估算了蒽晶体中分子间的电荷转移积分,并由此预测了蒽的空穴载流子迁移率为1.84 cm2· V-1·s-1.Kukhta等[10]研究了蒽类衍生物的离子势、电子亲合势和分子重组能等性质.目前对于蒽类衍生物的载流子传输性质,特别是蒽类衍生物的分子结构如何影响迁移率的理论研究还不是很深入,报道也很少见.
本文以三种蒽类衍生物为主要研究对象,分析影响三个化合物电荷传输的因素,寻找电荷的传输路径,并通过计算其重组能和传输积分,预测室温条件下的迁移率,并与同样方法计算得到的蒽的载流子迁移性质作比较进行分析,为这三种蒽类衍生物的实际应用提供理论基础.
1 理论基础与计算方法
对于有序的有机半导体材料的电荷传输,在低温下采用准能带的模型,通过有效质量的方法来计算空穴和电子的迁移率.而在室温或更高温度条件下,温度的升高带来热致无序化效应和散射,使能带的模型不再适用,通常采用电荷在分子间跳跃的机制来描述[4,9,11-12].
由Einstein方程式可知,迁移率μ与扩散系数D的关系可以表示为[9]
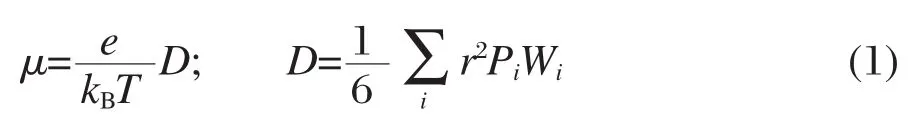
式(1)中,T为室温(298 K),e为电子电荷,kB为玻尔兹曼常数,D为电荷以一个分子为起点向三维空间方向上的扩散系数,Pi是电荷向第i个相邻分子迁移的几率,为相邻两分子的质心距离, Wi是电荷向第i个相邻分子的迁移速率,用Marcus电荷转移理论表示为[13]:

式(2)中,V为传输积分,h为Planck常数,λ为分子的重组能.要得到较大的迁移率,就要求物质具有尽可能小的重组能和尽可能大的传输积分.
重组能λ主要由内重组能和外重组能两部分组成[14-17].在晶体状态或无定形薄膜下载流子传输的实验和理论研究表明,外重组能的贡献很小[18-19],本文对其忽略不计.分子的内重组能λ是从中性态到离子态的几何弛豫能和与之相反过程的弛豫能两部分之和,即
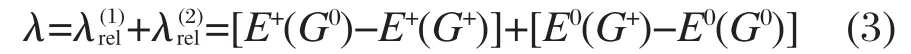
式(3)中,E+(G0)和E0(G0)为优化的中性分子构型下带一个正电荷和不带电荷时的能量,而E+(G+)和E0(G+)为优化的带正电荷的分子构型下带一个正电荷和不带电荷的能量.上式中E+和G+的上角标正号换成负号,就可以计算分子得到一个电子时的重组能.
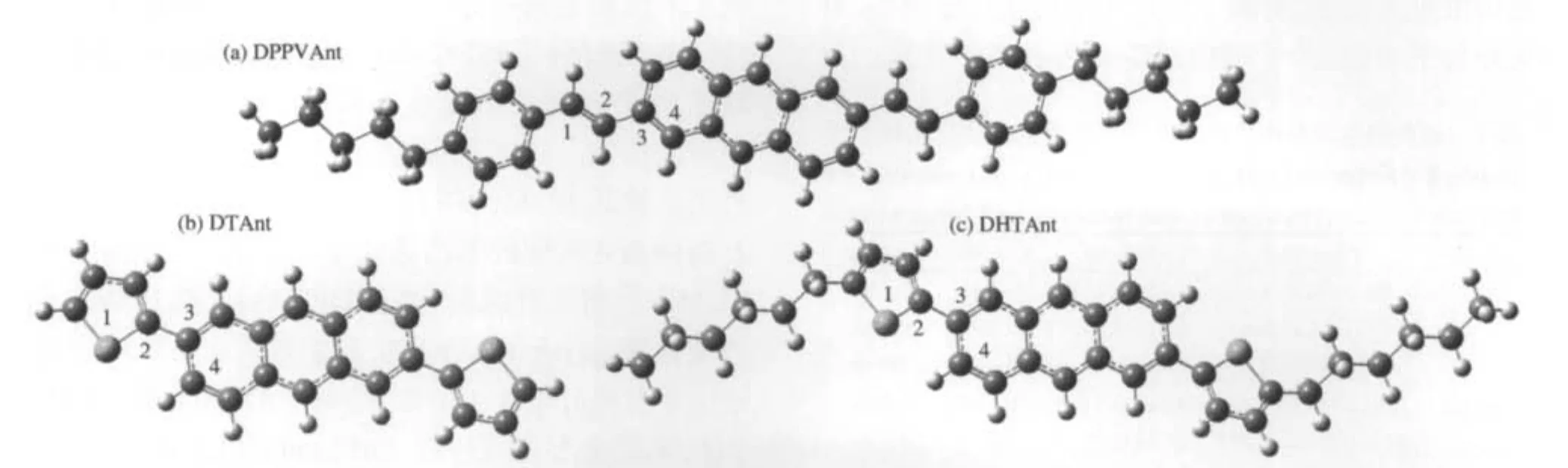
图1 三种蒽类衍生物中性态下优化的分子结构Fig.1 Optimized structures of the three anthracene derivatives in the neutral state
传输积分V的计算方法主要有:应用Koopman定理的间接法[20-22],直接耦合法[23-26],两态模型下的变分方法[27-29],分割技术方法[30-32],以及应用在光诱导电子转移体系的广义Mulliken-Hush方法[33-37]等.本文采用直接法求传输积分,可以用下式求算:

全部计算采用密度泛函理论[38-39]的B3LYP/6-31G(d)方法[40-41],用Gaussian 09软件包[42]完成.传输积分的数据处理采用自编的矩阵运算程序完成.
2 结果与讨论
2.1 分子结构和重组能
三种蒽类衍生物中性态的优化结构如图1所示.中性态、阳离子态和阴离子态的部分键长和二面角参数的计算值和实验值[7-8]见表1.计算得到的分子的重组能列于表2.与晶体结构相比,中性态的DPPVAnt中乙烯单元与蒽更趋于平面,而DTAnt和DHTAnt中的噻吩环与蒽具有更大的扭转角.这是由于晶体结构中分子排列受限的结果.与DPPVAnt中性态的优化结构相比,它的离子态的几何结构变化不大,C2—C3键长缩短0.002 nm,而C1—C2键长增长0.002 nm,阳离子的构型与中性态构型更为接近,阴离子的分子结构变化更大些,有更好的平面性.分子结构的变化与表2中计算的分子重组能是一致的.DPPVAnt的空穴和电子重组能分别为0.181和0.204 eV,空穴和电子重组能相近,而电子重组能稍大.而DTAnt和DHTAnt的离子态几何构型与中性态构型相比变化较大,并且阳离子构型要比阴离子构型变化大,尤其是噻吩环与蒽环的二面角,这些结构变化表明它们的空穴重组能会大于电子重组能,这也与计算的重组能结果是一致的,DTAnt和DHTAnt的空穴重组能大于电子重组能.计算得到的蒽类衍生物的空穴和电子重组能分别为0.137和0.198 eV,与文献的报道[10]吻合.三种蒽类衍生物的电子重组能相近,稍大于蒽的电子重组能,而空穴重组能均远大于蒽的空穴重组能,大小顺序为DPPVAnt<DTAnt<DHTAnt.仅从重组能对迁移率影响的角度来看,三种衍生物与蒽相比在重组能上对传输空穴没有优势.相比而言,化合物DPPVAnt具有较小的空穴重组能,更有利于空穴传输,而化合物DTAnt和DHTAnt不利于空穴传输.三种衍生物具有相近的电子重组能,在传输电子方面能力也应相近.影响蒽及其衍生物载流子的传输性能的另一因素是分子间的传输积分的大小,迁移率需要由这两个因素来共同决定.
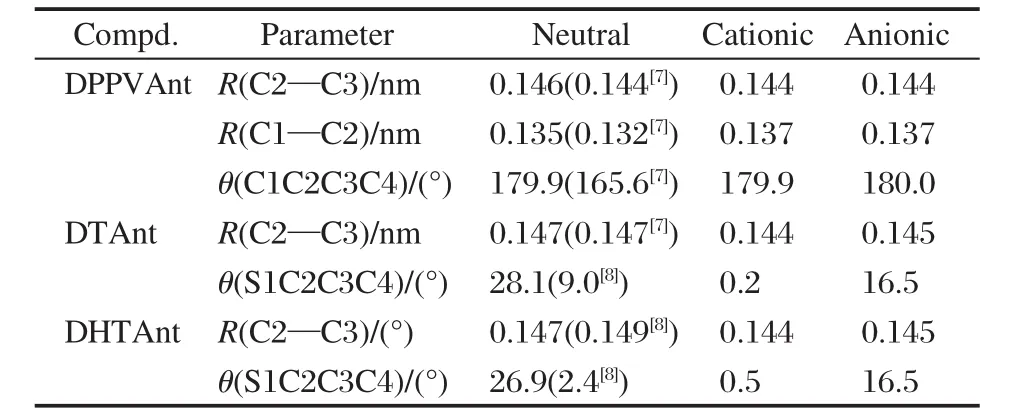
表1 蒽类衍生物在中性态和离子态下的部分优化结构参数Table 1 Selected optimized geometry parameters of anthracene derivatives in the neutral and ionic states
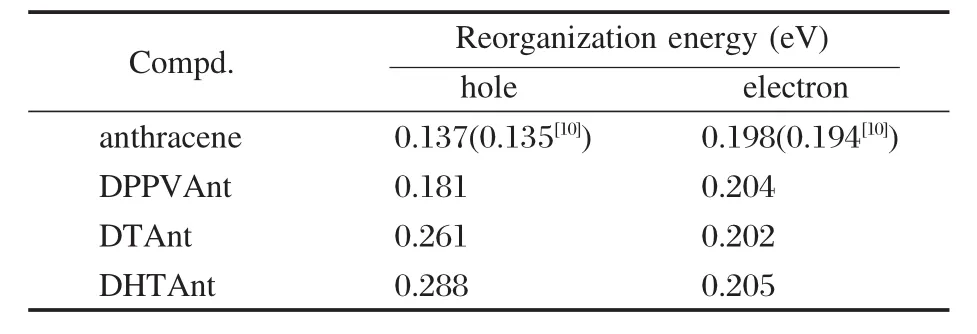
表2 蒽及其三种衍生物的空穴和电子重组能Table 2 Reorganization energies of anthracene and its three derivatives for hole and electron
2.2 分子的前线轨道
载流子传输与前线分子轨道分布密切相关[43],电子的传输是通过分子间的LUMO传递的,而空穴传输是通过分子间HOMO传递的.因而前线分子轨道电子云的分布以及离域程度的大小都会影响电荷的传输.三种衍生物的前线轨道如图2所示.三种衍生物的HOMO和LUMO几乎全部分布在蒽环及两端与之共轭的苯乙烯或噻吩环上,但HOMO和LUMO分布也有区别.对于DPPVAnt,其LUMO的离域性明显小于HOMO的,特别是两端的苯环上,电子云零散地分布在4个C上;对于DTAnt,其LUMO和HOMO分布比较接近.DHTAnt的LUMO的离域性会比DTAnt的大.DPPVAnt分子的离域长度最长,DHTAnt次之,DTAnt最短.从前线分子轨道的离域程度的角度可以预测,DPPVAnt的空穴传输能力会大于其电子传输能力;DTAnt很可能是较好的双极性传输材料,空穴和电子的传输会比较平衡;而DHTAnt相对于DTAnt会有利于电子的传输.

图2 三种蒽类衍生物中性态下分子的前线轨道图Fig.2 HOMOs and LUMOs plots of the three anthracene derivatives in the neutral stateHOMO:the highest occupied molecular orbital;LUMO:the lowest unoccupied molecular orbital
2.3 传输积分及迁移率
要计算分子间的电荷传输积分,必须确定电荷的传输路径.对于有机材料,在器件中主要以无定形态存在,随着实验手段的进步,薄膜越来越有序,在几十到几百纳米尺度下都可以看成是以有序的晶体形式进行堆积[44].不同的晶体结构就近似代表了薄膜的不同存在形态.我们以分子的晶体结构[7-8]确定分子在空间的相对位置,来计算分子的传输积分.选择晶体中的一个分子作为载流子的给体,该分子向周围每一个邻近分子的传输作为一种传输路径.三种蒽类衍生物和蒽分子中主要的传输路径如图3所示,计算得到的电子和空穴的传输积分值以及相应的计算和实验[7-8,45]迁移率见表3.
三种蒽类衍生物的晶体堆积密度都大于并五苯和蒽,其中DPPVAnt的堆积密度最大(结构参数a,b的大小列于表4,a,b方向见图3).三种蒽类衍生物中同层内的分子间距离参数a和隔层间距离参数b都比对应的并五苯[46]的小.与蒽相比,三种蒽衍生物中参数a与蒽的结构参数[47]相近,但参数b比蒽的小.DHTAnt的堆积也比DTAnt的堆积紧密.晶体呈现“人字形”排列,与蒽相似.DPPVAnt和DHTAnt的较大密度堆积是分子共轭芳香环之间π-π相互作用和两端烷基链之间较强的范德华相互作用力共同作用的结果.以DPPVAnt晶体为例,沿着分子长轴方向看,以一个分子为中心,与周围6个分子存在紧密的相互作用(见图3),其中4种为分子间等距离的分子边对面的“人字形”相互作用,为不同层间分子相互作用(如图4(a)所示).2种为同层分子之间等距离的错位平行相互作用(图4(b)所示).分子沿着长轴方向头尾相接的传输途径为8种,但是质心间距离较远,分子间相互作用较弱.所以分子中载流子的传输途径是沿着分子短轴方向为主,具有明显的各向异性的特点.
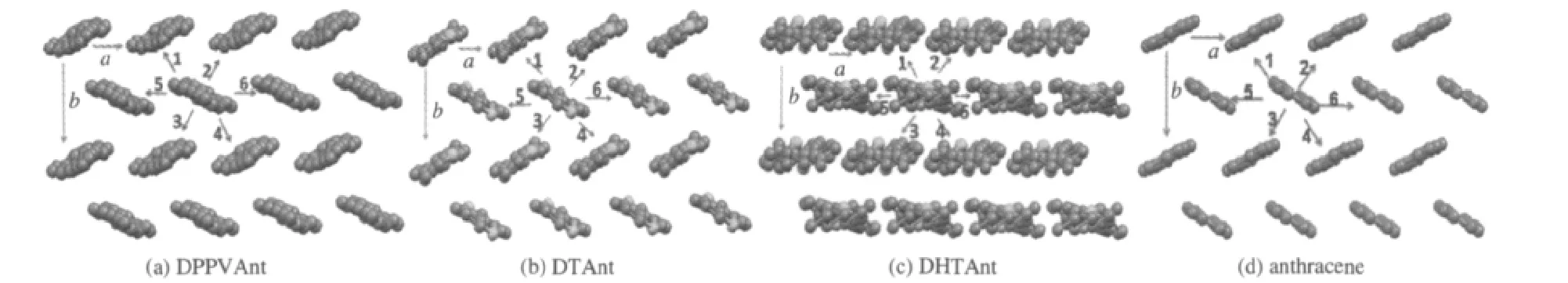
图3 蒽及其三种衍生物晶体结构中主要的传输路径Fig.3 Main pathways in the crystal structures of anthracene and its three derivatives
三种蒽类衍生物与蒽的电荷传输路径基本相同.以DPPVAnt为例,两种主要的传输路径中的轨道相互作用如图4所示.无论是“人字形”还是错位平行的排列中,空穴的相互作用都远大于电子的相互作用,电荷传输积分值见表3.利用式(1),室温下计算得到DPPVAnt的空穴和电子迁移率分别是0.49和0.042 cm2·V-1·s-1.空穴的载流子迁移率比电子的载流子迁移率约高一个数量级,表明DPPVAnt是较好的空穴传输材料.DTAnt中分子平行排列更有利于电子的传输,计算得到的空穴迁移率和电子迁移率分别为0.069和0.060 cm2·V-1·s-1,二者非常接近,表明化合物DTAnt可能是一种较好的双极性材料,载流子的传输比较平衡;而DHTAnt中两种分子排列方式中电子的传输积分都大于空穴的传输积分.计算的电子迁移率和空穴迁移率分别达到0.12和0.038 cm2·V-1·s-1,说明更有利于电子的传输.由此可见,不同的取代基可以改变蒽的传输性质,如DPPVAnt是以传输空穴为主,而DHTAnt是以传输电子为主.计算得到的蒽的空穴和电子的迁移率分别为0.25和0.22 cm2·V-1·s-1,比文献的计算值[45]小,可能是文献中采用两态模型计算会高估电荷转移积分,从而导致计算的迁移率偏大.与实验测得的总的载流子迁移率[7-8,45]相比,计算得到的迁移率数值与实验测量值处于相同的数量级内.计算得到的载流子迁移率能从更微观的角度理解材料的电荷迁移特性,例如是否更有利于电子或空穴传输.计算得出的载流子迁移率的规律与分子的重组能不一致,但与前线轨道的离域程度得到的结论基本一致.虽然蒽类衍生物的分子重组能远大于蒽的,但是蒽类衍生物的分子的堆积密度比蒽的大,从而传输积分的大小与蒽相当,计算得出的迁移率也与蒽在同样的数量级内.

表3 三种蒽类衍生物电子和空穴的传输积分和迁移率Table 3 Transfer integrals and mobilities of the three anthracene derivatives for hole and electron
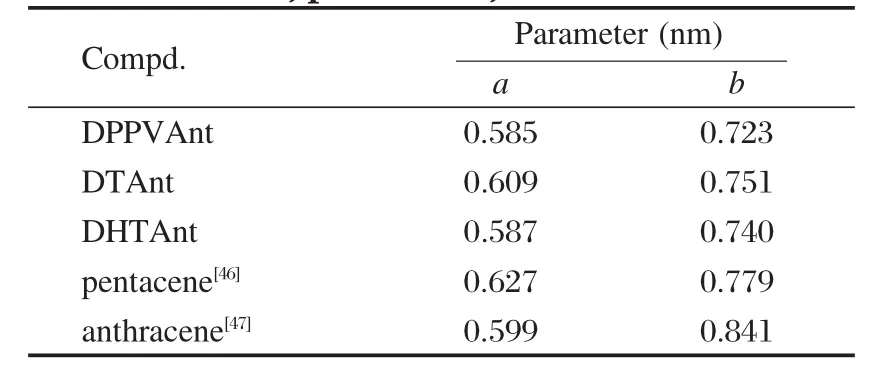
表4 蒽类衍生物以及并五苯和蒽的晶体结构参数Table 4 Crystal structure parameters of anthracene derivatives,pentacene,and anthracene

图4 DPPVAnt晶体的空穴和电子的两种主要传输路径中分子间轨道相互作用Fig.4 Intermolecular orbital interactions for two kinds of pathways of hole and electron in DPPVAnt crystal(a)herringbone mode;(b)displaced parallel mode
3 结 论
通过计算表明,蒽类衍生物具有与蒽和并五苯一样较高的载流子迁移率,计算与实验的结果相符. DPPVAnt是较好的空穴传输材料;DHTAnt有利于电子传输;而DTAnt是一种较好的双极性材料.同时可以看出,引入不同的取代基,在提高材料的稳定性的同时,还可以实现从传输空穴到传输电子的转变.三种蒽类衍生物的电子重组能相近,而空穴重组能均大于蒽的空穴重组能,其大小顺序为蒽<DPPVAnt<DTAnt<DHTAnt.这与计算的迁移率的结果不一致,说明分子的堆积结构决定了材料的电荷传输的性质.这类取代的蒽类衍生物由于具有较强的分子间相互作用,分子间堆积比较紧密,有利于载流子传输.
1 Pope,K.;Swenberg,C.E.Electronic processes in organic crystals and polymers.2nd ed.New York:Oxford University Press,1999
2 Silinsh,E.A.;Capek,V.Organic molecular crystals:interaction, localtion,and transport phenomena.New York:AIP Press,1994
3 Gershenson,M.E.;Podzorov,V.;Morpurgo,A.F.Rev.Mod. Phys.,2006,78:973
4 Coropceanu,V.;Cornil,J.;da Silva Filho,D.A.;Olivier,V.; Silbey,R.;Bredas,J.L.Chem.Rev.,2007,107:926
5 Shirota,Y.;Kageyama,H.Chem.Rev.,2007,107:953
6 Klauk,K.;Halik,M.;Zschieschang,U.;Schmid,G.;Radlik,W. J.Appl.Phys.,2002,92:5259
7 Meng,H.;Sun,F.P.;Goldfinger,M.B.;Jaycox,G.D.;Li,Z.G.; Marshall,W.J.;Blackman,G.S.J.Am.Chem.Soc.,2005,127: 2406
8 Meng,H.;Sun,F.P.;Goldfinger,M.B.;Gao,F.;Londono,D.J.; Marshal,W.J.;Blackman,G.S.;Dobbs,K.D.;Keys,D.E.J.Am. Chem.Soc.,2006,128:9304
9 Deng,W.Q.;Goddard III,W.A.J.Phys.Chem.B,2004,108: 8614
10 Kukhta,A.V.;Kukhta,I.N.;Kukhta,N.A.;Neyra,O.L.;Meza,E. J.Phys.B-At.Mol.Opt.Phys.,2008,41:205701
11 Yang,X.D.;Wang,L.J.;Wang,C.L.;Long,W.;Shuai,Z.G. Chem.Mater.,2008,20:3205
12 Wang,C.L.;Wang,F.H.;Yang,X.D.;Li,Q.K.;Shuai,Z.G. Organic Electrons,2008,9:635
13 Marcus,R.A.Rev.Mod.Phys.,1993,65:599
14 Marcus,R.A.J.Chem.Phys.,1965,43:679
15 Newton,M.D.;Sutin,N.Annu.Rev.Phys.Chem.,1984,35:437
16 Siders,P.;Marcus,R.A.J.Am.Chem.Soc.,1981,103:748
17 Brunschwig,B.S.;Logan,J.;Newton,M.D.;Sutin,N.J.Am. Chem.Soc.,1980,102:5798
18 Vilfan,I.Physica Status Solidi B-Basic Research,1973,59:351
19 Norton,J.E.;Bredas,J.L.J.Am.Chem.Soc.,2008,130:12377
20 Hutchison,G.R.;Ratner,M.A.;Marks,T.J.J.Am.Chem.Soc., 2005,127:16866
21 Lin,B.C.;Cheng,C.P.;You,Z.Q.;Hsu,C.P.J.Am.Chem.Soc., 2005,127:66
22 Cornil,J.;Beljonne,D.;Calbert,J.P.;Brédas,J.L.Adv.Mater., 2001,13:1053
23 Yang,X.D.;Li,Q.;Shuai,Z.G.Nanotechnology,2007,18: 424029
24 Troisi,A.;Orlandi,G.Chem.Phys.Lett.,2001,344:509
25 Yin,S.W.;Yi,Y.P.;Li,Q.X.;Yu,G.;Liu,Y.Q.;Shuai,Z.G. J.Phys.Chem.A,2006,110:7138
26 Gao,H.Z.;Qin,C.S.;Zhang,H.Y.;Wu,S.Y.;Su,Z.M.;Wang, Y.J.Phys.Chem.A,2008,112:9097
27 Liang,C.;Newton,M.D.J.Phys.Chem.,1992,97:3199
28 Dogonzdze,R.R.;Kuznetsov,A.M.;Vorotyntsev,M.A.Physica Status Solidi B-Basic Research,1972,54:425
29 Newton,M.D.Chem.Rev.,1991,91:767
30 Larsson,S.J.Am.Chem.Soc.,1981,103:4034
31 Löwdin,P.O.J.Mol.Spectrosc.,1963,10:12
32 Siddarth,P.;Marcus,R.A.J.Phys.Chem.,1990,94:2985
33 Hush,N.S.Electrochim.Acta,1968,13:1005
34 Creutz,C.;Newton,M.D.J.Photoch.Photobio.A,1994,82:47
35 Cave,R.J.;Newton,M.D.J.Chem.Phys.,1997,106:9213
36 Cave,R.J.;Newton,M.D.Chem.Phys.Lett.,1996,249:15
37 Kryachko,E.S.J.Phys.Chem.A,1999,103:4368
38 Hohenberg,P.;Kohn,W.Phys.Rev.,1964,136:B864
39 Kohn,W.;Sham,L.J.Phys.Rev.,1965,140:A1133
40 Becke,A.D.J.Chem.Phys.,1993,98:5648
41 Lee,C.;Yang,W.T.;Parr,R.G.Phys.Rev.B,1988,37:785
42 Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 09. Revision A.02.Wallingford,CT:Gaussian Inc.,2009
43 Liao,Y.;Su,Z.M.;Chen,Y.G.;Kan,Y.H.;Duan,H.X.;Qiu,Y. Q.;Wang,R.S.Chem.J.Chin.Univ.,2003,24:477 [廖 奕,苏忠民,陈亚光,阚玉和,段红霞,仇永清,王荣顺.高等学校化学学报,2003,24:477]
44 Shuai,Z.G.;Shao,J.S.Theretical chemistry:principles and applications.Beijing:Science Press,2008 [帅志刚,邵久书.理论化学:原理和应用.北京:科学出版社,2008]
45 Silinsh,E.A.;Capek,V.Organic molecular crystal:interaction, localization and transport phenomena.New York:AIP Press,1994: 332-333
46 Stefan,T.B.;Marta,M.T.;Peter,H.;Concepcioó,R.J.Am.Chem. Soc.,2004,126:6544
47 Brock,C.P.;Dunitz,J.D.Acta Crystallogr.Sect.B-Struct.Sci., 1990,46:795
February 22,2010;Revised:April 21,2010;Published on Web:June 11,2010.
Charge Transport Properties of Anthracene Derivatives
DUAN Gui-Hua1GAO Hong-Ze2WANG Li-Juan1ZHANG Hou-Yu1,*MA Yu-Guang1,*
(1State Key Laboratory of Supramolecular Structure and Materials,Jilin University,Changchun 130012,P.R.China;2Fundamental Department,Chinese People′s Armed Police Force Academy,Langfang 065000,Hebei Province,P.R.China)
The molecular geometries,electronic structures,reorganization energies,and charge transfer integrals of three anthracene derivatives{2,6-bis[2-(4-pentylphenyl)vinyl]anthracene,DPPVAnt;2,6-bis-thiophene anthracene, DTAnt;2,6-bis[2-hexylthiophene]anthracene,DHTAnt}were investigated by density functional theory at the B3LYP/ 6-31G(d)level.Their mobilities at room temperature were estimated using Einstein relations and compared with the calculated mobility of anthracene.DPPVAnt is a good hole-transporting material with a hole mobility as high as 0.49 cm2·V-1·s-1;DHTAnt is an electron-transporting material with an electron mobility of about 0.12 cm2·V-1·s-1;DTAnt is a bipolar material with its hole and electron mobilities being 0.069 and 0.060 cm2·V-1·s-1,respectively.The calculated mobilities were of the same magnitude as those obtained by experimental measurements.The reorganization energies for the electrons of the three derivatives are almost the same as that for anthracene but the reorganization energies for the holes of the three derivatives are larger than that of anthracene and they follow the order:anthracene<DPPVAnt<DTAnt<DHTAnt.This is not in agreement with the order of the calculated mobilities and implies that the mobilities are determined by molecular packing.
Density functional theory; Anthracene derivative; Mobility; Charge transport; Molecular reorganization energy
[Article] www.whxb.pku.edu.cn
*Corresponding authors.Email:houyuzhang@jlu.edu.cn,ygma@jlu.edu.cn.
The project was supported by the National Natural Science Foundation of China(20603013)and National Key Basic Research Program of China(973) (2009CB623605).
国家自然科学基金(20603013)和国家重点基础研究发展规划项目(973)(2009CB623605)资助
O641
猜你喜欢
物理学报(2022年6期)2022-03-30
车用发动机(2021年5期)2021-10-31
物理学报(2020年16期)2020-08-29
物理化学学报(2019年8期)2019-09-03
科技风(2018年9期)2018-05-14
小型内燃机与车辆技术(2016年4期)2016-10-21
浙江大学学报(工学版)(2016年2期)2016-06-05
烟草科技(2015年8期)2015-12-20
郑州大学学报(医学版)(2015年1期)2015-02-27
中央民族大学学报(自然科学版)(2014年4期)2014-06-09
