
meso取代卟啉衍生物的结构和光学性质
2010-11-30 10:56任雪峰任爱民封继康
物理化学学报 2010年1期
任雪峰 任爱民,* 王 钦 封继康,2
(1吉林大学理论化学研究所,理论化学计算国家重点实验室,长春 130023;2吉林大学化学学院,长春 130023)
meso取代卟啉衍生物的结构和光学性质
任雪峰1任爱民1,*王 钦1封继康1,2
(1吉林大学理论化学研究所,理论化学计算国家重点实验室,长春 130023;2吉林大学化学学院,长春 130023)
meso取代卟啉衍生物在红色电致发光材料上有较大的应用前景.本文采用密度泛函理论(DFT)B3LYP方法,对以反式二噻吩(S)作为能量传输供体的卟啉衍生物,Zn-5,10,15,20-tetra(2-[thiophen-2-yl]thiophene) porphyrin(SPZ)和5,10,15,20-tetra(2-[thiophen-2-yl]thiophene)porphyrin(TSP),进行了全优化.计算了二者的电离能(IP)、电子亲和势(EA)、空穴抽取能(HEP)、电子抽取能(EEP)、空穴和电子重组能(λ),评估了它们的载流子注入和传输能力.用含时密度泛函理论(TDDFT)/B3LYP/6-31G(d)方法计算了吸收光谱.用从头算单激发组态相互作用(CIS)方法优化了SPZ和TSP的最低激发单重态S1,并用含时Hartree-Fock(TDHF)方法研究它们的荧光光谱.理论计算结果表明,引入S基团对卟啉的光物理性质影响很大,尤其是电子注入和传输性质.
密度泛函理论;能量传输;载流子注入;传输能力
通常,红光材料是掺杂染料如四苯卟啉(TPP)[1]、铂卟啉[2]到具有较大能隙的主原料,通过空间能量传递(TS),把供体的激发能传递给受体,受体激发发射红光.但是,基于掺杂染料的红色有机光发射二极管材料的最优掺杂浓度很难控制[3,4],并且掺杂的工业制作过程复杂[5],因此满足人们要求的纯红光材料仍然很少.
最近,一系列以低聚芴为分支的卟啉衍生物被合成出来[6-10],卟啉和芴之间有化学键能量传递(TB).显然,TB比TS传递速度快[11],并且分支状卟啉可以有效地阻止荧光猝灭.Paul-Roth等[10]报道,从芴到卟啉的化学键能量传递效率高,并且这类化合物发射出饱和的红光同时具有高的发光效率,它的荧光量子产率比5,10,15,20-四苯卟啉(TPP)高[12].鉴于这种情况,我们研究并报道了这类卟啉衍生物的电子和光学特性[13].计算结果表明,这类衍生物具有显著的载流子注入和传输能力.显然,这种类型的化合物是潜在的红色发光材料.
这类分子的优越性质吸引我们进一步设计研究这类型的卟啉衍生物.用Turbomole程序包[14]中的含时密度泛函理论(TDDFT)和B3LYP/6-31G(d)方法计算得到S(2-[thiophen-2-yl]thiophene)的荧光发射光谱在356.5 nm.我们计算S的荧光光谱与其它理论计算值348 nm[15]和实验值362 nm[16]相吻合.并且,S基团的荧光发射光谱与芴的荧光光谱相近[10].因此,我们设计了Zn-5,10,15,20-tetra(2-[thiophen-2-yl] thiophene)porphyrin(SPZ)和5,10,15,20-tetra(2-[thiophen-2-yl]thiophene)porphyrin(TSP),其结构如图1所示,并研究它们的电子结构和电致发光性质.
1 计算方法
采用密度泛函理论(DFT)[17,18]/B3LYP[19-21]方法,在6-31G(d)基组下对SPZ和TSP的分子和离子几何构型进行了优化,所有分子采用C2h对称性限制.在优化几何基础上,计算电离能(IP)、电子亲和势(EA)、空穴抽取能(HEP)、电子抽取能(EEP)和重组能(λ).在基态平衡几何构型的基础上,用TD-B3LYP/ 6-31G(d)方法计算得到吸收光谱.用单激发组态相互作用(CIS)/3-21G(d)方法优化最低激发单重态S1的构型.在所得到的激发态几何基础上,用含时Hartree-Fock(TDHF)/6-31G(d)计算荧光光谱,这种方法在以前的工作中被验证适用于这类分子[13].所有计算都采用Gaussian 03程序包[22].
2 结果与讨论
2.1 基态几何结构

图1 由B3LYP/6-31G(d)方法优化的SPZ和TSP结构和标号示意图Fig.1 Optimized structures and atom label schemes of SPZ and TSP obtained at the B3LYP/6-31G(d)level
向卟啉Zn和卟啉的meso位置引入芴取代基,即5,10,15,20-四芴锌卟啉(FPZ)[13]和5,10,15,20-四芴卟啉(TFP)[13],有较好的发光性能.为提高这类卟啉衍生物的电子传输能力,我们向meso位置引入S取代基,即Zn-5,10,15,20-tetra(2-[thiophen-2-yl]thio-phene) porphyrin(SPZ)和5,10,15,20-tetra(2-[thiophen-2-yl] thiophene)porphyrin(TSP).优化的几何结构和标号列于图1,主要的键长和键角列于表1.如表1所示, SPZ和TSP的几何结构分别和文献中的FPZ和TFP的结构相似[13].如SPZ的键长Ca—Cb,Ca—N1和Ca—Cm与FPZ相比较偏离了0.0001 nm;而SPZ的键角α(CaCmCp)和α(CpN2Cp)与FPZ的相比较偏离小于0.5°.同样的,TSP的键长Ca—Cb,Ca—N1和Ca—Cm和TFP的相比较偏离了0.0001 nm;TSP的键角α(CaCmCp)和α(CpN2Cp)与TFP的相比较偏离小于5°.另外,Zn原子配位后,SPZ的Ca—Cb和Cp—N2键长比TSP的增长了0.0011 nm,而SPZ的Cp—Cq键长比TSP的减小了0.0016 nm.如图1侧面所示,卟啉环保持平面结构,而meso取代基S和卟啉环不共面.对于SPZ和TSP,S取代基团和卟啉环之间的环间角D(C1Cm′CmCa)分别是68.4°和66.2°,这主要是由与C1和Cb(或者Cq)相连接的H原子之间较强的排斥导致的.此外,较大的环间二面角使供体和受体保持各自性质独立,这也是能量传递的结构特点之一[23].
2.2 前线分子轨道
实验上,最高占据分子轨道(HOMO)和最低空分子轨道(LUMO)的能量是由Brédas提出的经验公式而获得的[24].对于材料的空穴和电子注入能力的初步判断,通常以HOMO与LUMO的能级是否与电极材料的功函数相匹配为依据.当空穴传输材料有较高的HOMO值时,说明它容易失去电子;当电子传输材料有较低的LUMO值,说明它容易接受电子.在本文中,HOMO和LUMO的能量由B3LYP/6-31G (d)计算得到的.SPZ和TSP的HOMO-1,HOMO, LUMO和LUMO+1的轨道能量绘于图2,前线分子轨道的电子云密度分布图绘于图3.
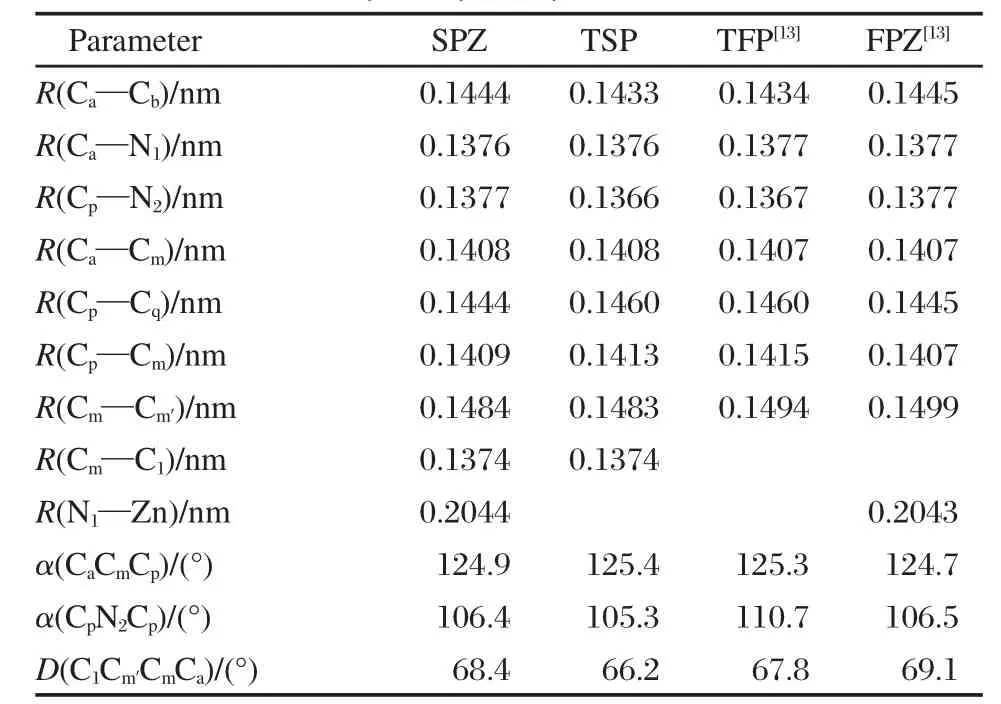
表1 分子SPZ,TSP,TFP和FPZ的主要结构参数Table 1 Selected structural parameters for molecules SPZ,TSP,TFP,and FPZ
如图2所示,SPZ的LUMO(-2.41 eV)比FPZ的LUMO能量值低,这表明SPZ的电子传输能力比FPZ强.同理,TSP的LUMO(-2.51 eV)比TFP的LUMO能量值低,这也表明TSP的电子传输能力比TFP强.而SPZ和TSP的HOMO能量都分别比FPZ和TFP低,这意味着SPZ和TSP的空穴传输能力比FPZ和TFP低.但是,由于SPZ和TSP的HOMO能量值和氧化铟锡(ITO)的功函值(-4.8 eV)[25]接近,说明SPZ和TSP也具有好的空穴传输能力.
如图3所示,SPZ和TSP的HOMO轨道上的电子云密度集中在S取代基和卟啉上,整体上表现出成键特性,而取代基和卟啉之间存在反键.它们的HOMO-1,LUMO和LUMO+1轨道上的电子云主要分布在卟啉环内,说明S取代基对这些分子轨道的贡献很小.
2.3 载流子的注入和传输能力
空穴和电子的注入和传输能力是衡量电致发光器件性能的重要参数,在这节里,我们计算了IP和EA值,分别用于衡量注入空穴和电子的难易.通常,空穴传输层(HTL)的IP值越低,空穴越容易从ITO注入到HTL;电子传输层(ETL)的EA越高,电子越容易从阴极注入到ETL.

图2 SPZ,FPZ[13],TSP和TFP[13]的HOMO-1,HOMO, LUMO和LUMO+1的轨道能量Fig.2 HOMO-1,HOMO,LUMO,LUMO+1 energies of SPZ,FPZ[13],TSP and TFP[13]

图3 SPZ和TSP的前线分子轨道电子云密度分布图Fig.3 Electron density distributions of frontier molecular orbitals of SPZ and TSP
用DFT方法计算得到的SPZ和TSP的IP,EA, HEP和EEP被列于表2.其中,垂直电离能IP(v)为在中性分子几何构型下阳离子和分子的能量差;绝热电离能IP(a)为中性分子和阳离子几何结构下的能量差;同样的,得到垂直电子亲和势EA(v)和绝热电子亲和势EA(a).而HEP和EEP分别是在阳离子和阴离子几何构型下抽取或失去一个电子的能量.
如表2所示,IP值依次降低:SPZ>TSP≈FPZ>TFP,这表明SPZ和TSP的空穴注入能力分别比FPZ和TFP要低.而EA值依次升高:FPZ<TFP<SPZ<TSP,这表明SPZ和TSP具有较高的电子注入能力.显然以上的研究结果和2.2节HOMO和LUMO能量预测的结论相一致.
根据我们以前的研究[13],载流子的传输能力用λ来衡量.有效的载流子传输应具有较小的重组能,并且空穴重组能λhole和电子重组能λelectron应该保持平衡.λhole和λelectron计算方法如下:

通过公式(1)和(2),我们计算得到了λhole和λelectron,并将结果列在表2.如表2所示,SPZ和TSP的λhole都比TFP和FPZ的大,显然TSP和SPZ的空穴传输能力比TFP和FPZ大.而SPZ和TSP的λelectron和FPZ和TFP的很相近,并且它们比广泛使用的电子传输性材料Alq3(λelectron=0.276 eV)[26]的重组能小,这表明从λ的角度看,SPZ和TSP可以作为好的电子传输材料.
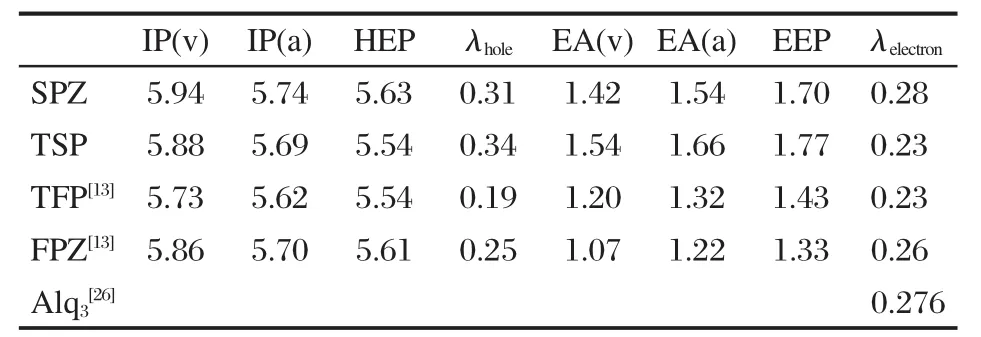
表2 研究分子的电离能、电子亲和势、抽取能和重组能Table 2 Ionization potentials,electron affinities, extraction potentials,and reorganization energies for studied molecules
2.4 电子吸收光谱和荧光光谱
TDDFT方法计算得到的电子吸收光谱模拟图绘于图4.如图4所示,SPZ和TSP的Q带在550-600 nm范围内,而它们的B带在450 nm处.值得注意的是,对于SPZ和TSP,在300-400 nm范围内有很强的宽峰,它覆盖了S取代基的荧光光谱(356.5 nm),这表明受体(SPZ和TSP)的吸收光谱和供体(S)的荧光光谱重叠,这也是TB型能量传递分子的重要结构特征.
基于优化的S1激发态几何构型,用TDHF/6-31G(d)方法计算了SPZ和TSP的荧光光谱.计算得到的荧光波长、振子强度和主要组态列在表3中.如表3所示,SPZ的荧光发射峰(677.1 nm)与FPZ的发射峰(660.8 nm[13])相比红移了16.3 nm.而TSP的荧光发射峰(744.5 nm)比TFP的发射峰(730.4 nm)红移了14.1 nm左右.
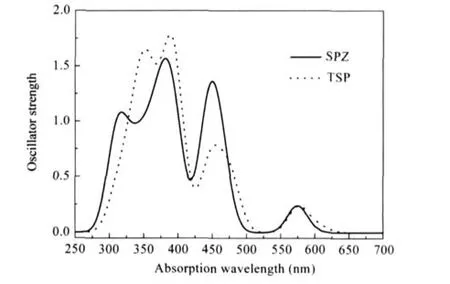
图4 由TD-B3LYP/6-31G(d)计算得到的SPZ和TSP的电子吸收光谱图Fig.4 Electronic absorption spectra of SPZ and TSP calculated at the TD-B3LYP/6-31G(d)level
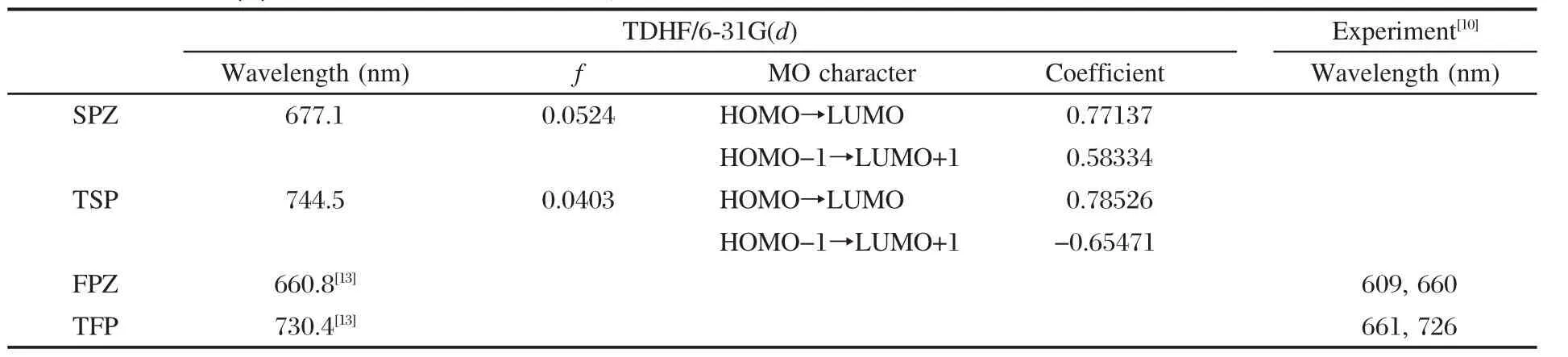
表3 由TDHF/6-31G(d)方法计算得到的SPZ和TSP的发射波长、振子强度(f)、主要跃迁轨道、跃迁系数以及FPZ和TFP[13]的荧光光谱性质Table 3 Emission wavelength,oscillator strength(f),dominant transition orbitals,coefficient calculated by TDHF/6-31G(d)method for SPZ and TSP,together with those properties of the compounds FPZ and TFP[13]
3 结论
本文研究了以(2-[thiophen-2-yl]thiophene)做能量传输供体的卟啉衍生物的结构和光电性质.理论计算结果表明,SPZ和TSP与文献中报道的分子FPZ和TFP有相似的几何结构.并且,SPZ和TSP有较大的环间二面角D(C1Cm′CmCa);受体(SPZ和TSP)的吸收光谱和供体(S)的荧光光谱重叠,这都是TB型能量传递分子的重要特征.值得注意的是,引入S基团, SPZ和TSP比FPZ和TFP有更好的电子注入和传输能力.
1 Virgili,T.;Lidzey,D.G.;Bradley,D.D.C.Adv.Mater.,2000,12: 58
2 Baldo,M.A.;O′Brien,D.F.;You,Y.;Shoustikov,A.;Sibley,S.; Thompson,M.E.;Forrest,S.R.Nature,1998,395:151
3 Bulovic,V.;Shoustikov,A.;Baldo,M.A.;Bose,E.;Kozlov,V.G.; Tompson,M.E.;Forrest,S.R.Chem.Phys.Lett.,1998,287:455
4 Hamada,Y.;Kanno,H.;Tsujioka,T.;Takahashi,H.;Usuki,T. Appl.Phys.Lett.,1999,75:1682
5 Chen,C.T.Chem.Mater.,2004,16:4389
6 Jiao,G.S.;Thoresen,L.;Burgess,K.H.J.Am.Chem.Soc.,2003, 125:14668
7 Strachan,J.P.;Gentemann,S.;Seth,J.;Kalsbeck,W.A.;Lindsey, J.S.;Holten,D.;Bocian,D.F.J.Am.Chem.Soc.,1997,119:11191
8 Li,B.S.;Li,J.;Fu,Y.Q.;Bo,Z.S.J.Am.Chem.Soc.,2004,126: 3430
9 Li,B.S.;Xu,X.J.;Sun,M.H.;Fu,Y.Q.;Yu,G.;Liu,Y.Q.;Bo, Z.S.Macromolecules,2006,39:456
10 Paul-Roth,C.O.;Simonneaux,G.C.R.Chim.,2006,9:1277
11 Speiser,S.Chem.Rev.,1996,96:1953
12 George,R.G.;Padmanabhan,M.Polyhedron,2003,22:3145
13 Ren,X.F.;Ren,A.M.;Feng,J.K.;Sun,C.C.J.Photochem. Photobiol.A-Chem.,2009,203:92
14 Ahlrichs,R.;Bär,M.;Baron,H.P.;Bauernschmitt,R.;Böcker,S.; Deglmann,P.;Ehrig,M.;Eichkorn,K.;Elliott,S.;Furche,F.; Haase,F.;Häser,M.;Horn,H.;Hättig,C.;Huber,C.;Huniar,U.; Kattannek,M.;Köhn,A.;Kölmel,C.;Kollwitz,M.;May,K.; Ochsenfeld,C.;Öhm,H.;Patzelt,H.;Rubner,O.;Schäfer,A.; Schneider,U.;Sierka,M.;Treutler,O.;Unterreiner,B.;von Arnim, M.;Weigend,F.;Weis,P.;Weiss,H.TURBOMOLE V5-7. Karlsruhe,Germany:Quantum Chemistry Group,University of Karlsruhe,2004
15 Liu,F.Y.;Zuo,P.;Meng,L.P.;Zheng,S.J.J.Mol.Struct.-Theochem,2005,726:161
16 Seixas de Melo,J.;Burrows,H.D.;Svensson,M.;Andersson,M. R.;Monkman,A.P.J.Chem.Phys.,2003,118:1550
17 Hohenberg,P.;Kohn,W.Phys.Rev.B,1964,136:864
18 Kohn,W.;Sham,L.J.Phys.Rev.A,1965,140:1133
19 Becke,A.D.Phys.Rev.A,1988,38:3098
20 Becke,A.D.J.Chem.Phys.,1993,98:5648
21 Lee,C.;Yang,W.;Parr,R.G.Phys.Rev.B,1988,37:785
22 Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, Revision B.04.Pittsburgh,PA:Gaussian Inc.,2003
23 Jiao,G.S.;Thoresen,L.H.;Kim,T.G.;Haaland,W.C.;Gao,F.; Topp,M.R.;Hochstrasser,R.M.;Metzker,M.L.;Burgess,K. Chem.Eur.J.,2006,12:7816
24 Brédas,J.L.;Silbey,R.;Boudreaux,D.S.;Chance,R.R.J.Am. Chem.Soc.,1983,105:6555
25 Jia,W.L.;Feng,X.D.;Bai,D.R.;Lu,Z.H.;Wang,S.; Vamvounis,G.Chem.Mater.,2005,17:164
26 Lin,B.C.;Cheng,C.P.;You,Z.Q.;Hsu,C.P.J.Am.Chem.Soc., 2005,127:66
August 2,2009;Revised:October 16,2009;Published on Web:November 10,2009.
Structural and Optical Properties of meso-Substituted Porphyrin Derivatives
REN Xue-Feng1REN Ai-Min1,*WANG Qin1FENG Ji-Kang1,2
(1State Key Laboratory of Theoretical and Computational Chemistry,Institute of Theoretical Chemistry,Jilin University, Changchun 130023,P.R.China;2College of Chemistry,Jilin University,Changchun 130023,P.R.China)
meso-substituted porphyrin derivatives show great potential for use as red light-emitting materials.We used density functional theory(DFT)with the B3LYP method to optimize the porphyrin derivatives Zn-5,10,15,20-tetra(2-[thiophen-2-yl]thiophene)porphyrin(SPZ)and 5,10,15,20-tetra(2-[thiophen-2-yl]thiophene)porphyrin(TSP) with the 2-[thiophen-2-yl]thiophene(S)group as an energy transport donor.Based on the optimized molecular structures, the ionization potentials(IP),electron affinities(EA),hole extraction potentials(HEP),electron extraction potentials (EEP),as well as hole and electron reorganization energy(λ)were calculated to investigate the charge injection and transport properties.We used the time dependent density functional theory(TDDFT)/B3LYP//6-31G(d)method to calculate the electronic absorption spectra of SPZ and TSP.Then the lowest excited singlet state(S1)of SPZ and TSP were optimized by the ab initio configuration interaction singlets(CIS)method.The fluorescence spectra of SPZ and TSP were calculated by the time dependent Hartree-Fock(TDHF)method.These theoretical calculations indicated that the introduction of the S groups significantly affected the photophysical properties of the porphyrin,especially the electron injection and transport properties.
Density functional theory;Energy transfer;Charge injection;Transport ability
O641;O644
*Corresponding author.Email:aimin.ren@gmail.com;Tel:+86-431-88499856.
The project was supported by the National Natural Science Foundation of China(20673045,20973078).
国家自然科学基金(20673045,20973078)资助项目
猜你喜欢
北京航空航天大学学报(2022年8期)2022-08-31
高中数理化(2022年2期)2022-02-22
车用发动机(2021年5期)2021-10-31
世界农药(2019年3期)2019-09-10
物理化学学报(2019年8期)2019-09-03
中学生数理化·高二版(2016年3期)2016-12-26
小型内燃机与车辆技术(2016年4期)2016-10-21
天然产物研究与开发(2016年11期)2016-06-15
中国光学(2015年5期)2015-12-09
食品工业科技(2014年23期)2014-03-11
