
芬太尼类化合物与阿片μ受体相互作用的分子对接与分子动力学模拟
2010-11-30 10:56:24胡文祥
物理化学学报 2010年1期
李 博 刘 明 胡文祥,*
(1首都师范大学物理有机与药物化学研究所,北京 100048;2首都师范大学生命科学学院,北京 100048)
芬太尼类化合物与阿片μ受体相互作用的分子对接与分子动力学模拟
李 博1刘 明2胡文祥1,*
(1首都师范大学物理有机与药物化学研究所,北京 100048;2首都师范大学生命科学学院,北京 100048)
采用分子对接和分子动力学(MD)模拟方法研究了芬太尼类化合物与阿片μ受体的相互作用机制.先用AutoDock4.0程序将芬太尼类化合物对接到同源模建的阿片μ受体结构中,再用GROMACS程序包在水溶液体系中分别对12个芬太尼激动剂和阿片μ受体蛋白复合物进行了MD模拟研究,优化对接复合物的结构,最后利用MM-PBSA方法,在APBS程序中计算芬太尼类衍生物与阿片μ受体的结合自由能,计算出的受体配合物结合常数(Ki)与其实验值吻合较好,并预测了化合物的活性排序.结果表明,复合物蛋白结构与空载受体蛋白结构有较大差异,特别是胞内区IL2、IL3和跨膜区段TM4骨架构象变化较大,不同的化合物对受体结构影响也有差异,活性较好的化合物会增加蛋白特定区域结构的柔性.芬太尼类化合物可能是通过和受体结合后诱导阿片μ受体构象转变为活性构象,引起一系列的信号传导激活G蛋白,从而引发生理效应.
分子动力学;芬太尼;阿片μ受体;分子对接
阿片μ受体是阿片受体μ、κ、δ三种重要亚型之一[1],目前这三种阿片受体的基因均已被克隆[2],研究发现阿片类选择性激动剂激动脊上和脊髓μ受体能引起一系列生理效应[3],具体表现为止痛效应、犒赏效应、心率减慢、呼吸抑制、肠蠕动抑制、僵住症和成瘾性等[4].吗啡为经典的阿片μ受体激动剂,纳洛酮为其拮抗剂[5].阿片μ受体属G蛋白偶联受体(G protein couple receptor,GPCR),GPCR具有相同的基本结构,即有一个细胞外氨基端区域,七个跨膜域以及一个细胞内羟基端尾区[6].
芬太尼(fentanyl)学名为1-苯乙基-4-(N-丙酰苯胺)哌啶,为阿片类镇痛药,它的镇痛效力约为吗啡的100-180倍,哌替啶的550-1000倍[7],并具有毒性低、对循环影响小、时效短(15-30 min)、容易控制、术后自主呼吸恢复快等优点,故近年来越来越受到人们的重视.芬太尼、舒芬太尼和阿芬太尼等芬太尼类配体药效增强、起效迅速、作用消失快,静脉滴注容易控制止痛剂量、安全可靠[8].关于芬太尼类激动剂与阿片μ受体选择性结合的分子作用机制的详细研究报道不多,目前对芬太尼活性构象的报道[9]中发现,芬太尼结构中的N-苯乙基和N-苯基基团的取向对活性构象贡献较大.本文采用芬太尼及本课题组设计的小分子配体与阿片μ受体蛋白进行分子对接和分子动力学模拟,以研究配体与蛋白间的疏水作用、氢键作用等关系,探讨其分子作用机制,对辅助设计新型高效镇痛剂和其他特殊用途的化合物有一定的意义.
1 模型和计算方法
1.1 蛋白结构来源
我们前期的研究结果与文献报道基本一致[10],故阿片μ受体蛋白的三维结构采用Zhang等[11]用同源模建方法搭建的结构.该结构以具有高解析度的视紫红激酶的X射线晶体结构为模板(PDB代码为:1F88).一般的GPCR受体经过同源建模后需要进行分子动力学模拟优化,目前,GPCR受体的MD模拟方法有两种,一种是先构建蛋白与脂水膜体系,然后用MD研究整个蛋白-膜-水体系.另一种是构建蛋白-水体系,主要用来研究配体结合性[12].本文采用后一种方法.
1.2 复合物结构对接与配体最优构象
在IBM IntelliStation POWER工作站上,采用AutoDock 4.0程序[13]对12个芬太尼受体激动剂小分子与阿片μ受体进行分子对接,对接参数:受体大分子的格点盒子大小为8 nm×8 nm×8 nm,格点间距为0.0375 nm,盒子中心位于受体中心,运用Lamarckian遗传算法,将局部能量搜索与遗传算法相结合,以半经验势函数作为能量打分函数,对小分子构象和位置进行全局搜索,对每个配体进行128次独立的对接实验.遗传算法程序关键参数为ga_pop_size 300 ga_num_evals 2500000,ga_run= 100,最后依据最低对接能和成簇分析的情况,选取合理的受体配体结合模式作为初步的复合物结构.
1.3 分子动力学模拟后再次对接
将MD模拟后得到的受体再次与配体进行对接,复合物的结构取MD达到稳态后的平均构象,然后从复合物结构中提取出受体结构和配体结构,再以新得到的受体按1.2节中的方法进行分子对接操作.
1.4 复合物结构的分子动力学研究
用GROMACS程序包[14]分别在水溶液体系中对12个芬太尼受体激动剂和阿片μ受体蛋白受体复合物进行了分子动力学模拟研究.12个复合物结构为1.2节所述对接的结果.每个复合物的质心位于立方体盒子中心,选择溶质原子到盒子壁的距离为0.7 nm,然后加入水分子,水分子选用SPC模型.采用GROMACS全原子力场,用2 fs积分步长,用最陡下降法进行了1000步的能量优化,然后在300 K下进行了10000步的限制性动力学优化,最后采用2 fs的步长,使用热浴耦合使系统温度保持在300 K,用Maxwell分布产生.每隔100步记录一次轨迹.在MD模拟中,用PME(Particle-Mesh Ewald electrostatics)方法处理静电作用,范德华相互作用截断半径取0.9 nm,用SUNWAY计算机集群并行计算模拟1200 ps,直到能量达到稳态为止.分别对蛋白环境和溶剂环境下激动剂与环境之间相互作用能进行系综平均,从而计算出受体-配体结合自由能(ΔG)和结合常数(Ki).
1.5 连续介质模型MM-PBSA方法计算复合物结合自由能
采用了MM-PBSA(molecular mechanics Poisson-Boltzmann surface area)方法[15]计算复合物结合自由能,此方法采用分子力学和连续介质模型估算复合物的结合自由能,并采用修正后的Robert Yang的Perl脚本[16]处理构象,修正后的脚本排除了程序中读取错误.
MM-PBSA方法的计算方程式:
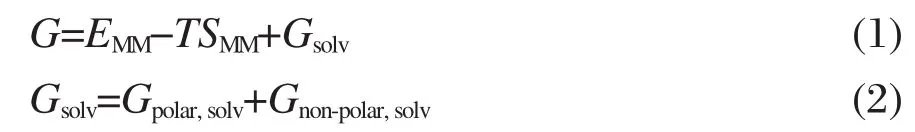
式(1)可以分为气相能量项和溶剂能量项,气相能量项包含内能项(EMM)和熵部分(TSMM)项;溶剂能量项Gsolv可以分为极性(Gpolar,solv)和非极性(Gnon-polar,solv)项,见式(2).用MD模拟后提取能量文件中的蛋白-蛋白电性和van der Waals相互作用而得到EMM项.参考了文献中的方法[17]考虑了在对接研究中不同复合物的熵相同而忽略了熵部分TSMM项.本文根据分子动力学结果,计算复合物加权协方差矩阵再计算其熵部分数值[14].极性和非极性项的计算则采用APBS软件包[18],其电性项的参数grid-spacing取0.01 nm.使用GROMOS96 43a1力场参数设置原子电荷和半径,探针半径0.14 nm,复合物介电常数设为1,溶剂的介电常数设为80[19].非极性项Gnon-polar,solv采用溶剂可及表面(SSASA)方法计算:

其中γ=2.2 kJ·mol-1·nm-2,β=3.84 kJ·mol-1[20].每个复合物采用21个结合构象,选取方法如下:在全部1200 ps模拟中,选取最后200 ps为平衡状态,即从1000 ps开始(包含1000 ps)取样,间隔为10 ps,选取一个结构,至1200 ps结束,共21个构象[21],采用这21个构象的极性和非极性项的平均值作为计算值.
2 结果与讨论
2.1 阿片μ受体蛋白结构特点
人类阿片μ受体蛋白有七个跨膜(TM)区段,属于GPCR超家族.该受体由400个氨基酸残基组成, TM2、TM3、TM7区和第2胞内环区(IL2)具有较高的同源性,而TM1、TM4、TM5区则同源性较低.第2胞内环和第3胞内环区(IL2和IL3)是蛋白结合区域,这些区域在三种阿片受体中的高度相似性表明有与蛋白相互作用的可能性.阿片μ受体与κ、δ两种阿片受体结构的最大差异部分在氨基端、羧基端及第2、3胞外环区(EL2和EL3),这些区域可能是阿片受体配体结合区,是不同配体选择性的结构基础.该推测已经通过直接点突变得到证实[22].阿片μ受体三维结构见图1.
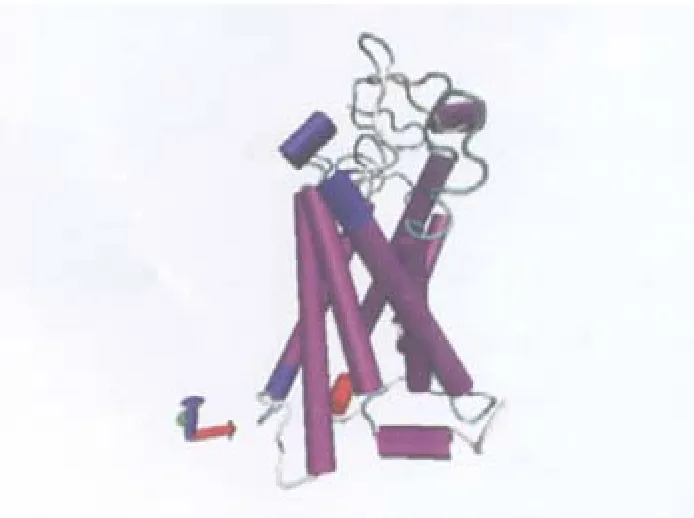
图1 阿片μ受体蛋白结构Fig.1 Protein structure of μ opioid receptor

图2 芬太尼类衍生物与μ阿片受体对接构象Fig.2 Docking conformation of fentanyl analogs with μ opioid receptor
2.2 复合物对接及结合能的计算
通过对初步的分子对接结果分析,发现芬太尼配体分子间距离0.5 nm内的参与配体相互作用比较重要的氨基酸有TM1区段上的Asp114、Ala117;TM3区段上的氨基酸Ile144、Ser145、Asp147、Tyr148、Asn150、Met151;TM5区段上的氨基酸His226、Ile230、Tyr234;TM6上Trp293、Asn296、His297、Tyr300、Arg303;TM7区段上的氨基酸Cys321、Ile322、Gly325、Tyr326等,所有参加作用的氨基酸编号见表1.12个芬太尼类衍生物(包括已知活性在内的4个芬太尼类衍生物)和阿片μ受体蛋白的对接结果有多种取向和构象,按照对接能量排序,我们发现低能构象多集中于配体质子化的N原子正电中心,并与TM3上的Asp147电负性中心发生相互作用(如图2所示),此外还有几个主要的作用残基,分别为TM1的Asp114,TM3的Asp147,和TM6的His297,其中Asp147与芬太尼类衍生物的哌啶季氨正电荷有电性作用,这些研究结果与现有文献的报道[24]是一致的.
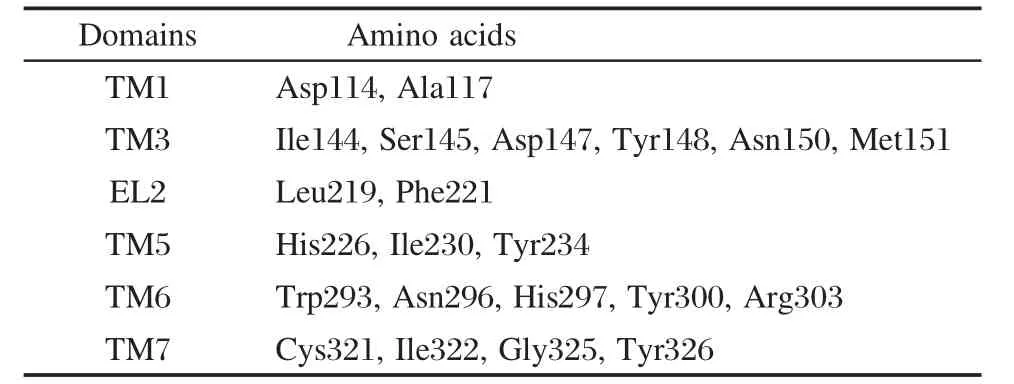
表1 各跨膜区段活性位点氨基酸Table 1 Amino acid of active pocket in transmembrane helices

图3 羟甲芬太尼通过水分子的氢键介导的相互作用Fig.3 Interaction of ohmefentanyl with watermediate through extra-H-bond
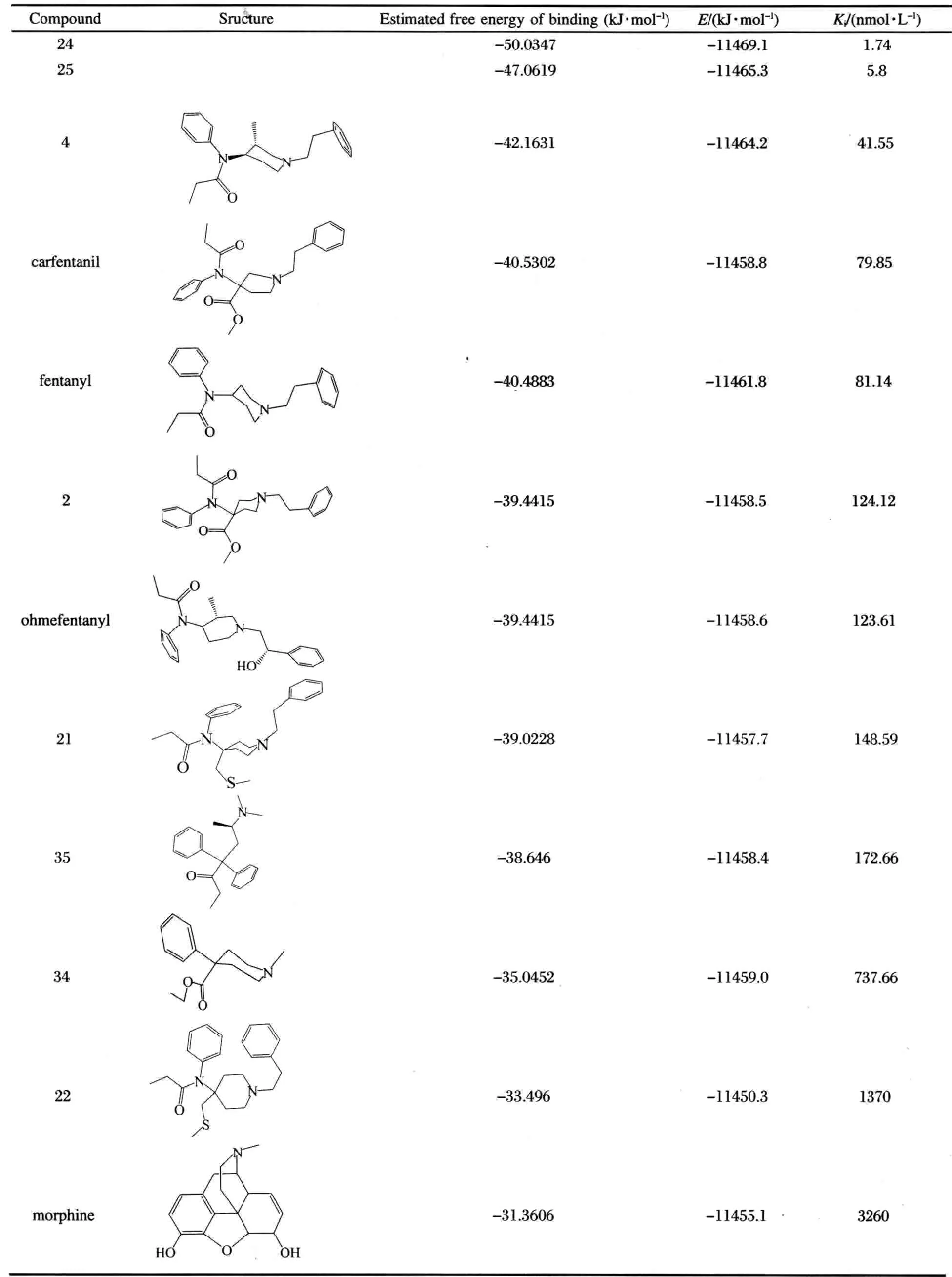
表2 芬太尼及类似物与MD模拟后受体的再对接结果Table 2 Re-docking results of fentanyl analogs after MD simulation
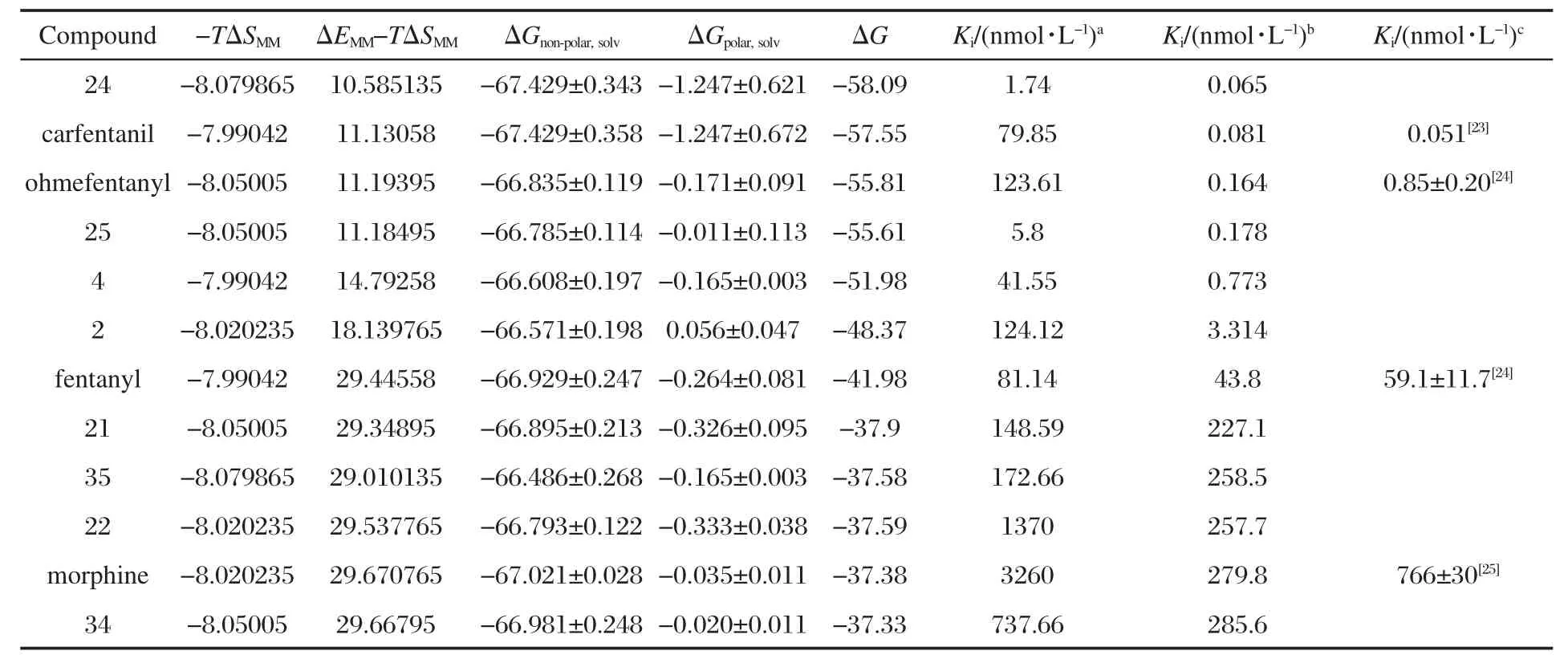
表3 芬太尼衍生物与阿片μ受体相互作用自由能Table 3 Free energy of interaction for fentanyl analogs and μ-opioid receptor complex
通过MD模拟研究,我们还发现复合物蛋白结构与无配体受体结构有较大差异(图2).因此,我们将MD模拟后得到的受体结构再次与各自配体进行分子对接研究,AutoDock 4.0再次对接后的计算结合能结果见表2.表2计算结果显示,结合能除羟甲芬太尼(ohmefentanyl)外,其它与实验活性排序一致.我们分析认为,羟甲芬太尼结构中额外的羟基通过水分子介导与Tyr148残基形成氢键(见图3),而在对接过程中这个溶剂分子引入的额外的氢键未给予考虑,从而羟甲芬太尼对接评分排序与实验结果是不一致的.采用MM-PBSA方法,计算出的12个芬太尼类衍生物的结合自由能,以及与它们对接计算的结果对比见表3.
用MM-PBSA方法计算得到的结合自由能,不但能够对抑制剂的结合强弱进行正确的排序,而且计算得到的数值和实验数值能够较好地符合.其中以芬太尼的计算结果最为接近,与本文前述AutoDock 4.0计算结果[13]相比较,MM-PBSA方法计算结果更为接近实验值,但计算数据与实验值仍略有偏差,除羟甲芬太尼外,计算值整体稍微偏高.
2.3 空载受体与复合物的MD模拟
2.3.1 空载受体的MD模拟
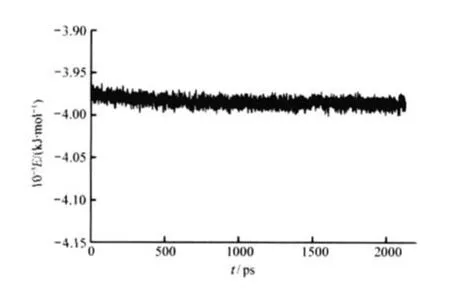
图4 受体在2000 ps范围的内能变化Fig.4 Internal energy fluctuation of the receptor in 2000 ps
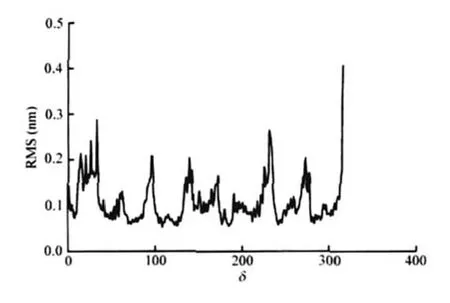
图5 受体蛋白Cα位置的序列位置(δ)的RMS变化Fig.5 RMS fluctuation with the position of sequence number(δ)for receptor αRMS:root mean square
空载受体蛋白在MD模拟过程中均方根偏差(RMSD)值是衡量体系是否稳定的重要依据,α碳(Cα)和骨架的RMSD值在初始的100 ps内变化大,随后基本稳定.蛋白结构经过2000 ps的优化达到稳态.本课题中以空载受体蛋白在1200 ps平衡后的平均构象作为分子对接的基础构象,研究了其内能的变化,如图4所示.结果表明,体系的内能变化不大,在短期模拟后即达到稳定状态,这可能是因为采用的初始结构已经进行了能量优化的原因,上述模拟结果表明我们所得系统已经达到了稳定状态. MD模拟中的Cα空间位置均方根(RMS)涨落是反映分子内部运动特征及柔性的一个重要参数,RMS值越高,柔性越大,否则相反.从图5所示的空载受体RMS图可以看出,全部序列中有7个跨膜区段(分别以TM1-TM7标示)柔性相对胞内(标示为IL)比胞外区(标示为EL)低,其TM4、TM5跨膜区氨基酸在平衡态时仍有一定的柔性.受体蛋白的柔性可用Cα序列位置(δ)的RMS变化来衡量,即观察蛋白的δ在模拟后与模拟前结构的变化,用位置偏离的RMS均方根偏差来衡量,如果δ的RMS变化较大,说明这些区段的柔性较大.如图6所示.
2.3.2 复合物模拟结果及不同配体复合物模拟结果差异

图6 受体蛋白骨架在2000 ps内的RMSD变化Fig.6 RMSD change of receptor protein backbone in 2000 psRSMD:root-mean-square deviation

图7 受体配体复合物在MD模拟期间能量(a)和骨架碳的RMSD(b)的变化Fig.7 Energy(a)and RMSD fluctuation(b)of receptor-liqand of complex C backbone in MD simulation
如图7所示,以芬太尼受体复合物为例的所有复合物分子的模拟,总能量和势能都很快达到稳态状态.在开始的500 ps内,RMSD变化稍大,500 ps后通过RMSD值变化可知,已基本达到平衡状态(图7(b)).这说明复合物体系已经稳定.分析复合物的RMS数据(图8)发现,受体柔性较大的片段为TM1、TM4、TM5跨膜区段,而对活性口袋具有主要贡献的TM2、TM3、TM6、TM7跨膜区段,结构柔性较低,比较稳定.
将芬太尼复合物蛋白部分在1200 ps内的Cα与空载配体受体结构Cα比较得知,复合物跨膜区段IL2、IL3(胞内区段)和TM4区段构象变化较大.分析复合物的RMS变化(图8、图9),发现结合配体后,受体TM1、TM3、TM6、TM7区段柔性降低(图8),而IL2、IL 3(胞内区段)EL3(胞外区段)和TM4的部分区段柔性增加,这说明配体能够稳定受体TM1、TM3、TM6、TM7的构象,通过改变增加部分区段构象和柔性来激活受体,配体对受体结构的影响不仅仅在活性区,可能还通过一系列构象和柔性变化影响到整个受体的功能,从而使蛋白转变为活性构象而发挥作用.我们前期通过构效关系和受体分子药理学研究[26-29]表明,受体蛋白活性中心的变化影响生理活性的大小,最可能仅对刚性受体而言是正确的.本研究表明,对大多数柔性受体来说,非活性中心构象的变化也将影响整个受体的活性构象及其与药物分子的相互作用,影响到药物分子的生物活性.

图8 芬太尼复合物的RMS变化Fig.8 RMS fluctuation of fentanyl complex

图9 芬太尼复合物与受体骨架碳的RMS变化比较Fig.9 Comparison with RMS fluctuation of the fentanyl complex and receptor backbone C

图10 对接评分高的复合物骨架碳的RMS变化Fig.10 RMS fluctuation of complex backbone C for high docking score complex
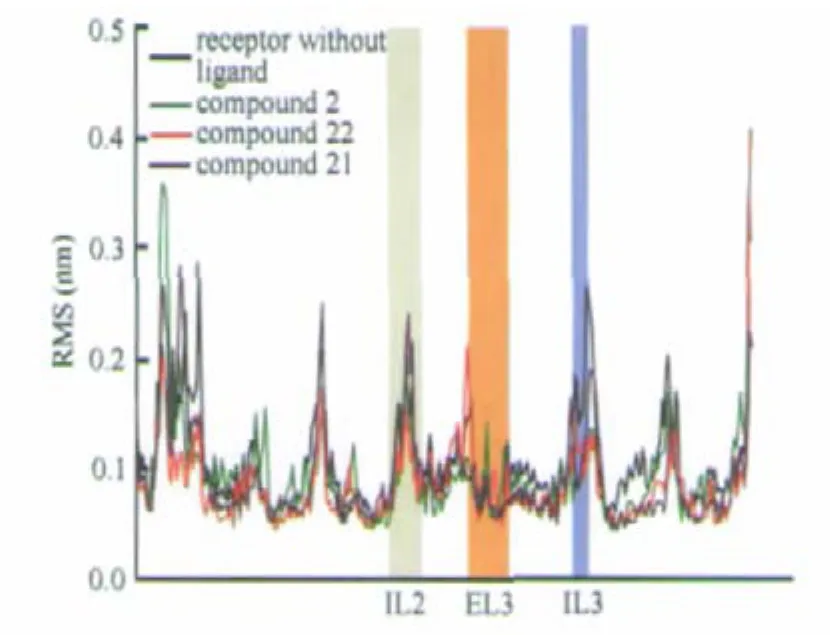
图11 对接评分低的配体与受体结合后的RMS变化Fig.11 RMS fluctuation of low docking score after ligand binding receptor
不同配体的复合物结构RMS变化有差异. Carfentanil 24结合受体后,受体的TM6、TM7区段RMS变化小,结构更加稳定,而IL2、IL3、EL3、TM4部分区段的RMS较高(如图10所示),实验值较好的化合物在稳定TM6、TM7区段跨膜蛋白结构的同时,还增加了其他蛋白区段的柔性,这些区段柔性和构象的改变可能与蛋白功能有关.
与受体结合能较低的配体2、21、22与受体结合后,受体各个区段的RMS变化都呈下降趋势,柔性降低(图11).这种变化说明配体2、21、22与受体结合后,使得受体结构柔性整体降低,但是由于TM4、IL2、IL3柔性可能与活性构象有关,柔性整体降低不利于蛋白向活性构象转变从而降低了活性.
2.3.3 结合配体后螺旋区的结构变化与蛋白活性构象
突变数据和分子对接的结果都表明TM2,TM3, TM7跨膜区段都参与了同配体的结合,因此有必要重点分析构成活性口袋的螺旋区结构变化.如图12所示,分析结合配体后受体螺旋区的RMSD的变化,发现结合配体后受体TM4、TM5的RMSD波动较其他的几个螺旋显著.
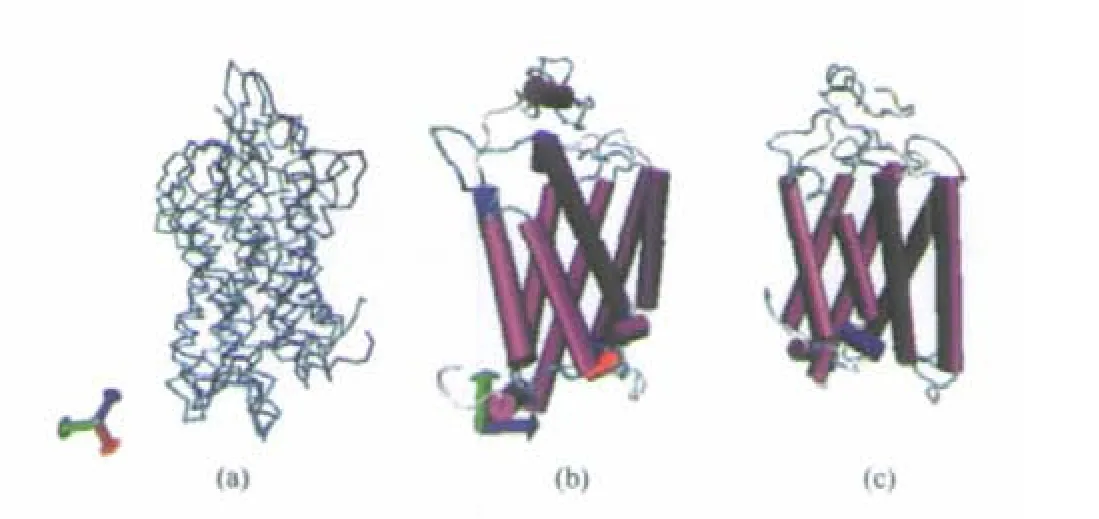
图12 受体与复合物蛋白结构的变化Fig.12 Change of structure with receptor and complex proteinstructures overlapping between receptor and complex(a), receptor(b),and complex(c)

表4 各跨膜区段结合配体后氢键数量的变化Table 4 Change of hydrogen bonds of transmembrane helices after binding with ligand
我们还分析了经历同样的MD模拟后,复合物和空载受体结构中氢键数量的变化,发现结合配体后,受体螺旋区的氢键除TM5区段外,其他区段的氢键数量稍有增加,特别是在TM4片段,比空载受体增加了两对氢键的相互作用,这与RMS变化分析中结合配体后引发TM4区段的变化相一致,而结合配体后整个体系氢键的相互作用增加,复合物与空载受体氢键数量比为63比55,见表4.
在空载受体TM1和TM7、TM2和TM7、TM2和TM3、TM5和TM6与TM6和TM7之间都有氢键的形成.经过分析,这些螺旋之间形成氢键的现象在G蛋白偶联受体家族中是普遍存在的.在结合配体之后,TM1和TM7、TM2和TM7、TM2和TM3、TM5和TM6跨膜区段之间的氢键作用消失,TM1和TM2、TM2和TM4跨膜区段之间的氢键作用增加,TM2、TM3、TM7是结合口袋所在的区域,我们推测这种氢键的消失和增加是受体活性构象所必须的.配体与Asp147、Tyr148、Ile322、Gly325、Tyr326等氨基酸残基之间发生的相互作用打破了原有螺旋片段之间的相互联系,引起几个TM区段之间发生较大程度的相对运动,这种氢键作用关系的重构可能是蛋白转变为活性构象的重要因素.在配体分子进入受体后,直接影响的是TM2、TM3、TM7跨膜区段,而随后影响的是TM4区段,这样使得下游的信号分子转变为活性的状态,引起一系列的信号变化而激活G蛋白,从而引发生理效应.
3 结论
对一系列芬太尼类化合物与阿片μ受体蛋白进行分子对接,讨论了各配体与μ受体进行对接后蛋白质的构象,在分子动力学模拟基础上,采用MM-PBSA方法进行了自由能排序,得出了已知活性分子的排序,具有比较好的预测能力,并与实验值较接近.
对复合物进行的分子动力学模拟研究发现,受体与小分子配体结合后影响了非口袋区的蛋白构象.从蛋白结构不同区段的RMS变化分析发现, TM4跨膜区段和EL1胞外区的构象改变较大,这是与传统活性构象不同之处.
通过探讨复合物与受体氢键的变化,发现与配体结合后,复合物氢键的数目较单一受体略有增加,原有的TM4区段的氢键关系有所变化,推测TM4区段氢键的变化可能是蛋白活性构象改变的重要因素.
用分子动力学模拟结果结合MM-PBSA方法计算自由能作为评分依据,能得到比较好的理论预测结果,这些为药物分子设计提供了重要的理论基础.
1 Piestrzeniewicz,M.K.;Michna,J.;Janeka,A.Postepy Biochem., 2006,52(3):313
2 Liang,Y.;Mestek,A.;Yu,L.;Carr,L.G.Brain Res.,1995,679: 82
3 Martin,W.R.;Eades,C.G.;Thompson,J.A.Pharmacol.Exp. Ther.,1976,197(3):517
4 Yamamoto,T.;Shono,K.;Tanabe,S.Pharmacol.Exp.Ther., 2006,318(1):206
5 Brownstein,M.J.Proc.Natl.Acad.Sci.U.S.A.,1993,90:5391
6 Dhawan,B.N.;Cesselin,F.;Raghubir,R.Pharmacol.Rev.,1996, 48:567
7 Liu,J.J.;Zhao,J.Modern anesthesiology.Beijing:People Health Press,1995:222 [刘俊杰,赵 俊.现代麻醉学.北京:人民卫生出版社,1995:222]
8 Rosow,C.E.Anesth.Analg.,1999,89:81
9 Subramanian,G.;Ferguson,D.M.Drug Des.Discov.,2000,17 (1):55
10 Morris,G.M.;Goodsell,D.S.;Halliday,R.S.;Huey,R.;Hart,W. E.;Belew,R.K.;Olson,A.J.J.Comput.Chem.,1998,19:1639
11 Zhang,Y.;Sham,Y.Y.;Rajamani,R.;Gao,J.L.;Portoghese,P.S. Chem.Bio Chem.,2005,6:853
12 Hess,B.;Bekker,H.;Berendsen,H.J.C.J.Comput.Chem.,1997, 18:1463
13 Srinivasan,J.;Cheatham,T.E.;Cieplak,P.J.Am.Chem.Soc., 1998,120:9401
14 http://cholla.wustl.edu/baker/classes/BME-540/2008/mm-pbsa/
15 Wu,Y.;Cao,Z.;Yi,H.Biophys.J.,2004,87:105
16 Baker,N.A.;Sept,D.;Joseph,S.Proc.Natl.Acad.Sci.U.S.A., 2001,98:10037
17 Ganother,A.;Friedman,R.;Nachliel,E.Biophys.J.,2006,91:2436
18 Chong,L.T.;Duan,Y.;Wang,L.Proc.Natl.Acad.Sci.U.S.A., 1999,96:14330
19 Chen,Y.;Mestek,A.;Liu,J.Mol.Pharmacol.,1993,44:8
20 Humphrey,W.;Dalke,A.;Schulten,K.J.Mol.Graphics,1996,14 (1):33
21 Dosen-Micovic,L.J.Bioorg.Med.Chem.,2006,14:2887
22 Surratt,C.K.;Johnson,P.S.;Moriwaki,A.J.Biol.Chem.,1994, 269:20548
23 Frost,J.J.;Wagner,Jr.H.N.;Dannals,R.F.J.Comput.Assist. Tomogr.,1985,9(2):231
24 Liu,Z.H.Acta Pharmacologica Sinica,2003,24(9):864
25 Bartlett,S.E.;Smith,M.T.Life Sciences,1995,57(6):609
26 Hu,W.X.;Wang,J.Y.Synergy combination chemistry.Beijing: Science Press,2003:1-71 [胡文祥,王建营.协同组合化学.北京:科学出版社,2003:1-71]
27 Zhang,Z.Y.;An,L.Y.;Hu,W.X.;Xiang,Y.H.J.Comput.Aided Mol.Des.,2007,21:145
28 Hu,W.X.;Yun,L.H.Chin.Sci.Bull.,1994,39(10):856
29 Zhu,H.W.;Fang,H.;Wang,L.Y.;Hu,W.X.;Xu,W.F.Drug Discov.Ther.,2008,2(1):192
May 8,2009;Revised:September 8,2009;Published on Web:November 25,2009.
Molecular Docking and Molecular Dynamics Simulations of Fentanyl Analogs Binding to μ-Opioid Receptors
LI Bo1LIU Ming2HU Wen-Xiang1,*
(1Institute of Physical Organic and Medicinal Chemistry,Capital Normal University,Beijing 100048,P.R.China;2College of Life Science,Capital Normal University,Beijing 100048,P.R.China)
Weperformedmoleculardockingandmoleculardynamics(MD)simulations to investigate the interactions between fentanyl analogs and μ-opioid receptors.The AutoDock 4.0 program was used to perform the docking and homology modeling of the μ-opioid receptor structure.MD method as implemented in the GROMACS program was used to model the twelve fentanyl receptor agonists and the μ-opioid receptor protein compounds in water and to optimize the docking complex structure.Based on MM-PBSA methods,the APBS program was used to calculate the binding affinity of the complexes and the binding contant of receptor and liqand(Ki)values determined using MMPBSA were consistent with the experimental values.Our predictions of compound activity sequences were,therefore, correct.The MD simulations of these complexes revealed that the protein structures in the complexes differed substantially from the structures of the ligand-free receptors.The backbone of the intracellular region segments IL2, IL3 and TM4 showed that the skeleton conformations had changed significantly.Different compounds may influence the receptor structure differently.Compounds with high activities may enhance binding flexibility in certain protein structural regions.These facts imply that fentanyl analogs may result in μ-opioid receptors changing to an active conformation after receptor binding.Physiologic effects may thus be triggered by a mediating signal transduction and by the activation of the G-protein.
Molecular dynamics; Fentanyl; μ-opioid receptor; Molecular docking
O643
*Corresponding author.Email:huwx66@163.com;Tel:+86-10-68904756.
The project was supported by the National Natural Science Foundation of China(20872095).
国家自然科学基金(20872095)资助项目
猜你喜欢
中学生理科应试(2024年1期)2024-05-18 13:02:52
中国药理学与毒理学杂志(2017年4期)2017-06-01 11:31:13
温州大学学报(自然科学版)(2016年1期)2016-10-27 14:57:48
湖南农业科学(2016年7期)2016-03-13 14:31:44
西南军医(2015年2期)2015-01-22 09:09:44
应用化工(2014年7期)2014-08-09 09:20:23
原子与分子物理学报(2014年3期)2014-02-28 22:18:23
无机化学学报(2014年5期)2014-02-28 17:31:40
无机化学学报(2014年1期)2014-02-28 17:30:06
食品科学(2013年13期)2013-03-11 18:24:39
