
G-CSF对肌萎缩侧索硬化细胞外谷氨酸摄取和抗自由基作用的影响
2010-09-20 08:03:40朴铁花蔡鸿彦席中原
中风与神经疾病杂志 2010年12期
朴铁花, 蔡鸿彦, 席中原, 卢 蕾, 宋 磊
谷氨酸是中枢神经系统内最主要的兴奋性递质。任何原因引起的突触间隙或细胞外谷氨酸的异样积累都会造成谷氨酸受体的过度兴奋从而引起神经细胞损害-兴奋性毒性作用[1]。肌萎缩侧索硬化症(amyotrophic lateral sclerosis,ALS)是特异性累及上下运动神经元的一种神经系统变性疾病,是最为常见的运动神经元疾病[2]。国内外学者通过对 ALS患者体内谷氨酸含量测定显示患者体内有兴奋性氨基酸的失衡,血中和脑脊液中谷氨酸含量较正常人明显升高,认为谷氨酸的兴奋性毒性作用可能是ALS发病的重要机制之一[3]。随后一系列对 ALS发病与 Glu的体内、体外研究发现,任何原因引起的细胞外谷氨酸清除障碍均可能导致脊髓前角运动神经元的损伤及缺失[4]。
除了氨基酸的兴奋性毒性,自由基的氧化作用在ALS发生发展中扮演着关键的角色[5]。Hall等应用SOD1/G93A转基因小鼠来检测脂质过氧化对脊髓的损害作用时发现运动神经元中丙二醛的含量异常增高,提示脂质过氧化是疾病进程中的一个重要方面[6]。进一步研究表明,运用环氧化酶抑制剂 COX-2拮抗剂SC236可明显减少脊髓运动神经元的脱失[7]。Henderson等给 ALS患者服用谷胱甘肽过氧化物酶前体(NAC),发现可提高 GPX的含量,减轻运动神经元的变性[8]。上述研究均显示了ALS抗氧化治疗的潜力。
发病机制的复杂性决定了 ALS治疗非常困难,目前被认为有效并上市的药品只有力鲁唑。但力鲁唑是一种谷氨酸的拮抗剂,只能通过减轻谷氨酸兴奋性毒性发挥作用[9,10]。为了弥补力鲁唑治疗 ALS的局限性,迫切需要寻找一种可以针对 ALS多种发病机制的临床新药物。粒细胞集落刺激因子(granulocyte colony stimulating factor,G-CSF)是一种分子量为19.6kD的糖蛋白,主要由单核/巨噬细胞分泌,在体内可以特异性调节粒细胞的增殖与分化,增强粒细胞功能[11]。目前研究表明,G-CSF还具有抗自由基和阻断谷氨酸兴奋毒性等抗凋亡作用[12,13],并可能对ALS的治疗有效[14]。本实验以脊髓切片体外培养构建 ALS模型,研究 G-CSF是否可以通过抗自由基和兴奋氨基酸毒性作用发挥对脊髓前角运动神经元的保护作用,为其进一步应用于治疗 ALS提供依据。
1 材料与方法
1.1 材料 8日龄 SD乳鼠由吉林大学动物实验中心提供。动物许可证号:SCXK(吉)2003-0002。MS培养液(50%MEM含 25mmol Hepes+25%马血清 +25%Hank平衡盐液含 25.6mg/ml葡萄糖)、GBSS(Geys平衡盐液含 6.4mg/ml葡萄糖)购自美国 Gibco公司,6孔培养板(Greiner bio-one公司),MilliporeMillicell-CM膜(美国 Millipore公司),BX 51光学显微镜(日本奥林巴斯),一抗(小鼠抗非磷酸化神经丝单克隆抗体)、二抗(生物素化马抗小鼠IgG)、SABC免疫组织化学试剂盒(武汉博士德公司 ),DAB(美国 Sigma),Glu、SOD、MDA测试盒(南京建成生物工程研究所),重组粒细胞集落刺激因子(长春金赛药业有限责任公司),McⅡWain组织切 片 刀 (Geneq),DL-threo-β-hydroxyaspartate、L-trans-pyrrolidine-2,4-dicarboxylate(Sigma)。
1.2 器官型脊髓(organotypic spinal cord)的建立与培养 按Rothstein和Corse所报道的方法:将 8日龄 SD乳鼠分别快速地在碘酒、75%酒精中各浸泡消毒后断头处死。迅速分离并取出整条脊髓放入GBSS中,在解剖显微镜下分离并剪断腰段脊髓的神经根,小心剥离脊膜。无菌条件下用 McⅡWain组织切片刀将腰段脊髓切成 350μm厚的薄片后,转移至 GBSS中室温下慢慢分离成单片。取 6孔培养板,每孔内放入 1ml MS培养液,并放置 Millipore-Millicell-CM多孔膜。用吸管将完好的脊髓片转移至膜上,每孔放置 5片,移去膜表面多余的培养液,入 CO2培养箱(36.5℃,5%CO2+95%空气),每 3d换 MS培养液 1次。
1.3 ALS模型的建立以及分组 10d后,将培养的脊髓切片从培养箱中拿出,随机分为 3组:对照组、ALS模型组和 G-CSF组,每组 6孔。对照组不做任何处理,仍常规培养;ALS组模型的建立按照 Matyja等[15]方法向每孔中加入 DL-threo-β-hydroxyaspartate(THA)至终浓度为 100μmol,以后加入 L-trans-pyrrolidine-2,4-dicarboxylate(PDC)至终浓度为 500μmol;G-CSF组在 ALS模型组处理的基础上再加入重组人粒细胞集落刺激因子(rhG-CSF)至终浓度 10ng/ml[16]。将各组重新放入培养箱内培养,每 3d换半液 1次。每次换液后 3组重复上述处理。
1.4 培养液中谷氨酸含量测定 培养 1w、2w、3w、4w时在每次换液前取培养液(每组 6份),按南京建成生物工程研究所提供的谷氨酸检测试剂盒进行测定,具体步骤如下:(1)取 0.2m l的培养液加0.6ml的试剂 1,充分混匀,离心 10min(3500r/min),取上清液待测。(2)设立空白管、标准管和测定管,在空白管中加入蒸馏水 0.5ml;在标准管中加入0.2mmol/L谷氨酸标准应用液 0.5ml;在测定管中加入待测上清液 0.5ml。然后在各管中加入 1ml试剂 1、0.1m l试剂 2、0.01ml试剂 4和 0.39ml蒸馏水。混匀后室温放置 10min,340nm处,1cm光径,蒸馏水调零,测各管吸光度 A1值。(3)在各管中再加入 0.02ml试剂 5,混匀,37℃水浴 40min,取出后于 340nm处,1cm光径,蒸馏水调零,测各管吸光度A2值。计算公式:谷氨酸浓度(μmol/L)=[测定管(A2值 -A1值)-空白管(A2值 -A1值)]÷[标准管(A2值 -A1值)-空白管(A2值 -A1值)]×标准管浓度(20μmol/L)×4。
1.5 脊髓取材 每组取 6孔(每孔 5片)脊髓组织切片,取右侧腹前角组织称重后,应用冰冷的生理盐水制备成10%组织匀浆置 -40℃待测。余脊髓组织浸泡于 4%多聚甲醛缓冲液,用于免疫组化检测。
1.6 脊髓前角中 SOD测定 根据南京建成生物工程研究所提供的 SOD检测试盒说明书进行脊髓前角中 SOD活性的测定和计算。测定步骤如下:(1)测定管中加试剂 1 lm l、10%组织匀浆 30μl、试剂 2 0.lm l、试剂 3 0.1ml、试剂 4 0.1m l;对照管中加试剂 1 1ml、试剂 2 0.1m l、试剂 3 0.1m l、试剂 4 0.1 m l。(2)用漩涡混匀器充分混匀,置 37℃恒温水浴40min。(3)测定管、对照管中分别加显色剂 2m l。(4)混匀,室温放置 10min,于波长 550nm处,1cm光径比色杯,蒸馏水调零,测各管 OD值。计算公式:SOD活力(U/mgp rot)=(对照管OD值 -测定管 OD值)÷对照管 OD值 ÷50% ×3.83÷0.03÷0.67mgpor/tml÷对照管 OD值 ×0.465。
1.7 脊髓前角中 MDA测定 根据南京建成生物工程研究所提供的 MDA检测试盒说明书测定脊髓前角中 MDA含量。测定步骤:(1)标准管中加10nmol/ml的标准品 0.1ml和试剂 1 0.1ml;标准空白管中加无水乙醇 0.1m l和试剂 10.1ml;测定管中加 10%脑组织匀浆 0.1ml和试剂 1 0.1ml;测定空白管中加 10%脑组织匀浆 0.1ml和试剂 1 0.1m l。(2)漩涡混匀器混匀,试管口用保鲜膜扎紧,用针头刺一小孔,95℃水浴 80min,取出后流水冷却,离心10min(4000r/min)后取上清,532nm下,1cm光径,蒸馏水调零,测各管 OD值。组织中 MDA含量(nmol/mgport)=(测定管 OD值 -测定空白管 OD值)÷(标准管 OD值 -标准空白管 OD值)×10nmol/ml÷蛋白含量(mgprot/nmol)。
1.8 免疫组化染色计数前角运动神经元 取各组以 4%多聚甲醛固定的脊髓切片,按 Iwasaki等[5]使用的方法进行脊髓前角运动神经元的免疫组化染色,具体步骤如下:将脊髓切片先用 0.1mol/L PBS冲洗 3次,10%马血清 /TBS阻断 60min,加入单克隆抗非磷酸化抗体 SMI-32(含 0.5%TritonX-100)后,放置于在 4℃摇床过夜。次日使用 TBS洗3次(每次 10min)后,加入二抗生物素标记的马抗小鼠 IgG(1∶1000),25℃下 60min后 TBS洗 3次(每次 10m in),加入辣根过氧化物酶标记的链酶卵白素(1∶200),25℃下 60m in后 TBS洗 3次(每次10min);DAB显色 3~10min,TBS洗 3次(每次10min);脱水、透明、封片。以 TBS代替一抗为阴性对照。位于脊髓的腹侧、SMI-32(+)、并且胞体直径大于 30μm的细胞被认定为 α运动神经元。每组取 6张切片,100倍镜下每张切片取 5个视野,对前角运动神经元计数并取其平均值。
1.9 统计学处理 应用 SPSS11.5 forWindows统计软件进行分析。本组数据均为计量资料,数据以均数 ±标准差(±s)表示,采用方差分析及 q检验。检验水准 α=0.05。
2 结 果
2.1 脊髓器官模型的形态学 培养 10d后的脊髓片在体外生长良好,体积逐渐增大,组织边缘有光晕。在相差显微镜下可见培养液清亮无混浊,脊髓切片的背角略暗,含有大量的密集的小体积细胞;脊髓腹角透光性较背角强,细胞大、多角而稀疏。HE染色可清楚显示脊髓前角运动神经元(见图1)。
2.2 培养液中 Glu浓度变化 结果显示对照组培养液中 Glu浓度较低,并且随时间变化(1w、2w、3w、4w)变化不大,基本维持在恒定水平;ALS模型组培养液中 Glu浓度随时间延长逐渐增加,并显著高于对照组;G-CSF组培养液 Glu浓度随时间延长逐渐增加,显著高于对照组,但在 2w后 Glu浓度显著低于 ALS模型组(见图2)。
2.3 脊髓前角中 SOD、MDA的含量 ALS模型组脊髓前角中 SOD活性显著低于对照组(P<0.05),G-CSF组 SOD、GSH-PX活性高于 ALS模型组(P<0.05),但仍低于对照组(P<0.05)。ALS模型组前角中 MDA含量显著高于对照组(P<0.01),G-CSF组 MDA含量低于 ALS模型组(P<0.05),但仍高于对照组(P<0.05)(见表1)。
2.4 脊髓前角运动神经元数量 免疫组化染色结果显示,对照组脊髓切片腹侧前角 α运动神经元分布稀疏,数量为 20±3.3,胞体呈多角形,深棕色,有细长突起,彼此间可见突触联系;ALS模型组α运动神经元极少,为 5±1.1,个别切片难以计数;G-CSF组脊髓切片 α运动神经元数量为 14±2.7,较对照组减少(P<0.05),但明显多于 ALS模型组(P<0.05)(见图3)。
表1 各组SOD和 GSH-PX活性数值(±s,n=6)

表1 各组SOD和 GSH-PX活性数值(±s,n=6)
与 ALS组相比*P<0.05
组别(酶) 对照组 ALS模型组 G-CSF组SOD活性(U/mgprot)MDA含量(nmol/mgprot)159.04±2.34 1.16±0.09 108.91±2.84 3.66±0.92 138.76±3.16*1.93±0.13*

图1 HE染色可清楚显示脊髓前角运动神经元,稀疏,呈深染(×40)
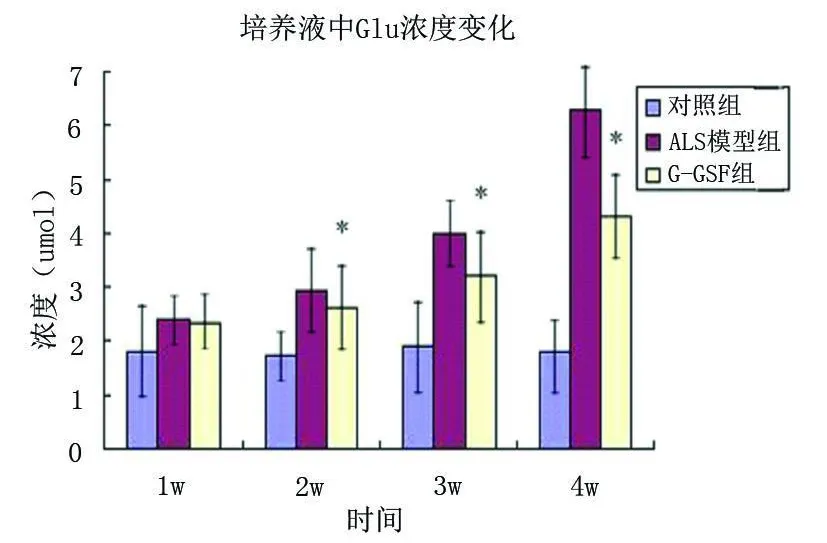
图2 示不同时间点各组培养液中 Glu浓度的变化
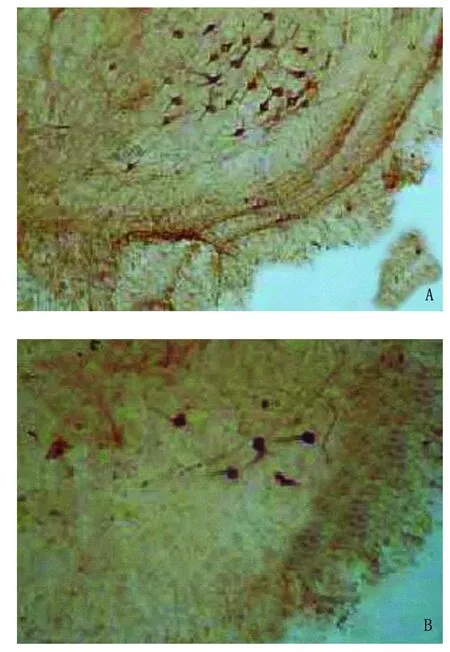

图3 A、B、C分别为对照组、ALS模型组和 G-CSF组,脊髓前角 SM-32染色(×100),可见G-CSF组脊髓切片 α运动神经元数量较对照组减少(P<0.05),但明显多于 ALS模型组(P<0.05)
3 讨 论
目前 ALS的实验模型研究主要有转基因动物模型、免疫介导的动物模型和体外细胞或组织培养模型。以 Cu/Zn SOD基因突变和神经丝过度磷酸化为基础的转基因小鼠模型主要针对发病率仅占 ALS 5%~10%左右的家族性 ALS,且对实验条件、设备、技术要求高,周期长,花费大。而免疫介导动物模型又不能精确地模拟 ALS的自然慢性发病过程。故本实验采用目前应用最为广泛的体外细胞(或组织、器官)培养模型对 ALS进行研究。在培养的脊髓切片中加入谷氨酸转运体拮抗剂 THA和 PDC,造成细胞外谷氨酸无法被摄取导致在培养液中浓度不断增加,从而选择性地损伤脊髓前角运动神经元,较好地模拟了 ALS的发病过程,并可缩短整个实验的周期,易操作,干预条件易调控,有利于进行下一步对 GCSF神经保护作用进行研究。
正常情况下,细胞外液中没有使谷氨酸失活的水解酶系统,将细胞外液谷氨酸快速摄取到细胞内是清除 Glu毒性的唯一途径,而这一作用是靠分布在谷氨酸能神经元末梢和神经胶质细胞膜上的谷氨酸转运体完成的[17]。实验显示,在对照组的脊髓器官模型中,加入的 Glu可以被正常清除而不会引起细胞毒性,培养液中的 Glu浓度始终维持在一个恒定的水平。而在加入谷氨酸转运体拮抗剂THA和PDC的 ALS模型组培养液中,Glu浓度在 1w时测定即明显高于对照组,随时间变化逐渐增加。先前的研究发现 G-CSF在体外实验中可以增加细胞外谷氨酸的摄取,在活体实验中可以明显减轻缺血后谷氨酸的释放[16,18,19]。在此次实验中我们发现 1w后,GCSF组培养液中 Glu的浓度明显低于 ALS模型组,说明 G-CSF在谷氨酸转运体被一定拮抗的情况下同样可以增加 Glu的摄取。我们认为 G-CSF这一作用是通过与谷氨酸能神经元和神经胶质细胞膜上的粒细胞集落刺激因子受体(G-CSF-R)相结合实现的。G-CSF-R被认为存在于神经元和胶质细胞上[20],GCSF与之结合后可激活 JAKs-STATs途径,通过上调基因增加谷氨酸转运蛋白的表达而减轻细胞外谷氨酸的浓度[21]。
同前所述,氧自由基在 ALS发病机制中起到核心作用,并且和 Glu的兴奋性毒性作用是密不可分的。Dawson等在原代神经细胞培养时,证实过量的NO有助于 Glu的毒性。而当胞外 Glu升高时,可以通过激活非 NMDA受体增加 Ca2+内流,使细胞内和线粒体内 Ca2+超载,造成电子溢出成为自由基,形成瀑布式链锁反应,对运动神经元造成不可逆性损害[22]。在我们先前对 G-CSF的研究中,发现其可以增强缺血再灌注后抗氧化酶系统的活性,降低脂质过氧化反应。在本次研究中,发现在 G-CSF组细胞中 SOD活性明显高于 ALS模型组,而 MDA水平明显下降。
最后我们还对所有的脊髓切片进行 SMI-32染色。SMI-32染色可以很好地显示神经系统神经元的胞体、树突和某些较粗大的轴突,目前已代替过去常用的抗乙酰胆碱酯酶抗体,作为脊髓运动神经元标记物。本实验根据文献认定脊髓的腹侧 SMI-32(+)并且胞体直径大于 30μm的细胞为 α运动神经元,发现在 G-CSF组脊髓前角的运动神经元数目明显多于 ALS模型组,证实了 G-CSF的神经保护作用可以通过增加细胞外 Glu的摄取和抗自由基作用实现,提示 G-CSF可能作为治疗 ALS的新药物值得进一步研究。
[1]Olney JW.Neurotoxicity of excitatory am ino acids[M].in:McGeer EG,Olney JW,McGeerPL(Eds.),Kainic Acid asa Tool in Neurobiology,Raven Press,New York:1978.95-121.
[2]Row land LP,Shneider NA.Medicalprogress:amyotrophic lateral sc lerosis[J].N Eng JMed,2001,344(22):1689-1700.
[3]IwasakiY,Ikeda K,KinoshitaN,etal.Plasmaamino acid levels in patients with amyotrophic lateral sc lerosis[J].JNeurol Sci,1992,107(2):219-222.
[4]Zagam iCJ,Beart PM,Wallis N,etal.Oxidative and excitotoxic insults exert differential effects on spinal motoneurons and astrocytic glutamate transporters:Implications for the role of astrogliosis in amyotrophic lateral sclerosis[J].Glia,2009,57(2):119-135.
[5]Doroudchi MM,Minotti S,Figlewicz DA,etal.Nitrotyrosination contributes minimally to toxicity of mutant SOD 1 associated with ALS[J].Neuroreport,2001,12(6):1239-1243.
[6]HallED,Andrus PK,Ostveen JA,etal.Relationship of oxygen radicalinduced lipidperoxidative damage todisease onset and progression in a transgenic model of familial ALS[J].JNeurosci Res,1998,53(1):66-67.
[7]Drachman DB,Rothstein JD.Inhibition of cyclooxygenase-2 protects motor neurons in an organotypicmodelofamyotrophic lateralsclerosis[J].Ann Neurol,2000,48(5):792-795.
[8]Henderson JT,JavaheriM,Kopko S,etal.Reduction of lower motor neuron degeneration in wobbler m ice by N-acetyl-L-cysteine[J].Neuroscience,1996,16(23):7574-7582.
[9]Bensimon G,Lacomblez L,Meininger V,etal.A controlled trial of riluzole in amyotrophic lateral sclerosis[J].New Engl JMed,1994,330:585-591.
[10]Lacomblez L,Bensimon G,Leigh PN,etal.Dose-ranging study of riluzole in amyotrophic lateral sclerosis[J].Lancet,1996,347(16):1425-1431.
[11]Ghristopher PH,Timothy O,Eisenberg D,etal.The structure ofgranulocyte colony stimulating factor and its relationship to other growth factors[J].Biochem istry,1993,90:5167-5171.
[12]Jung KH,Chu K,Lee ST,etal.G-CSF protects human cerebral hybrid neuronsagainstin vitro ischemia[J].Neurosci Lett,2006,394(3):168-173.
[13]Minnerup J,Sevimli S,SchabitzWR,etal.Granulocyte-colony stimulating factor for stroke treatment:mechanisms of action and efficacy in prec linical studies[J].Exp Transl Stroke Med,2009,1:2.
[14]Zhang Y,Wang L,Fu Y,etal.Preliminary investigation of effect of granulocyte colony stimulating factor on amyotrophic lateral sc lerosis[J].Amyotroph Lateral Scler,2009,10(5):430-431.
[15]Matyja E,Nagańska E,Taraszewska A,etal.Themode ofspinalmotor neurons degeneration in amodel of slow glutamate excitotoxicity in vitro[J].Folia Neuropathol,2005,43(1):7-13.
[16]Schabitz WR,Kollmar R,SchwaningerM,etal.Neuroprotective effect of granulocyte colony-stimulating factor after focal cerebral ischem ia[J].Stroke,2003,34(3):745-751.
[17]Oshea RD.Rolesand regulation ofglutamate transporters in the central nervous system[J].Clin Exp Pharmacol Physiol,2002,29(11):1018-1023.
[18]Han JL,Blank T,Schwab S,etal.Inhibited glutamate release by granulocyte-colony stimulating factor after experimental stroke[J].Neurosci Lett,2008,432(3):167-169.
[19]ChenWF,Sung CS,Jean YH,etal.Suppressive effects of intrathecal granulocyte colony-stimulating factor on excessive release of excitatory amino acids in the spinal cerebrospinal fluid of rats with cord ischem ia:role of glutamate transporters[J].Neuroscience,2010,165(4):1217-1232.
[20]Hintzen RQ,Voormolen J,Sonneveld P,etal.Glioblastoma causing granulocytosis by secretion of granulocyte colony-stimulating factor[J].Neurology,2000,54(1):259-261.
[21]Wu GJ,Chen WF,Sung CS,etal.Preventive effects of intrathecal methylprednisolone adm inistration on spinal cord ischem ia in rats:the role of excitatory amino acid metabolizing systems[J].Neuroscience,2007,147(2):294-303.
[22]Emerit J,Edeas M,Bricaire F,etal.Neurodegenerative diseases and oxidative stress[J].Biomed Pharmacother,2004,58(1):39-46.
猜你喜欢
中学生物学(2021年8期)2021-11-02 04:53:14
考试与评价·高二版(2020年2期)2020-09-10 07:22:44
中国果业信息(2019年1期)2019-01-05 17:41:42
中国畜牧兽医文摘(2018年6期)2018-07-28 02:30:16
三门峡职业技术学院学报(2017年1期)2017-06-05 10:17:30
中国洗涤用品工业(2017年2期)2017-04-16 05:07:45
中国比较医学杂志(2017年5期)2017-01-17 06:17:05
中外医疗(2015年16期)2016-01-04 06:51:42
实用手外科杂志(2015年1期)2015-08-27 01:52:06
科学启蒙(2015年8期)2015-08-07 03:54:46
