
局灶性缺血预处理对鼠脑 NF-κB表达及其靶基因 COX-2 mRNA转录的影响
2010-08-25 03:55郝玉曼罗祖明姜晓峰
中风与神经疾病杂志 2010年8期
郝玉曼, 罗祖明, 姜晓峰, 高 励, 曾 仲, 张 仲, 刘 艳
环氧化酶(cyclooxygenase,COX)又称前列腺素G/H合成酶,是花生四烯酸转化为前列腺素的限速酶,有结构型(COX-1)及诱导型(COX-2)2种亚型,其转录和表达受核因子-кB(nuclear factor-кB,NF-кB)的调控。业已证实,COX-2在肿瘤和炎症的发病中起到重要作用[1],新近研究发现,COX-2可能还参与了脑缺血再灌注损伤[2]。
越来越多的证据表明,短暂的、亚致死性脑缺血预处理(ischemic preconditioning,IPC)可诱导缺血耐受 (ischemic tolerance,IT)作用从而减轻其后发生的严重脑缺血所致的神经损伤[3],其分子机制尚不清楚,初步研究发现,NF-кB信号转导途径可能在 IT的产生中扮演了重要角色,但迄今为止,有关缺血预处理对 NF-кB/COX-2通路的影响,国内外尚未见报道。我们利用在前期工作中建立的脑缺血耐受模型,观察了局灶性脑缺血预处理对 NF-κB表达及其靶基因 COX-2 mRNA转录的影响。
1 材料与方法
1.1 动物分组 健康雄性 Sprague-Dawley大鼠共 36只,体重 250~320g,由四川大学实验动物中心提供,随机分为预缺血组、缺血组及对照组,每组各 12只。预缺血组(IPC+MCAO组):以 10min大脑中动脉阻塞(MCAO)作为 IPC,3d后再次给予 2h MCAO,再灌注 22h后处死。缺血组(SS+MCAO组):假手术 3d后给予 2h MCAO,再灌注 22h后处死。对照组(SS+SS组):两次均为假手术,在第 2次假手术后 24h处死。
1.2 缺血预处理模型制备 采用由 Longa法改进而来的大脑中动脉二次线栓法。大鼠用 10%水合氯醛(0.3ml/100g)腹腔注射麻醉,颈部正中切口,结扎颈外动脉(ECA)远端,用提拉线暂时阻断颈总动脉(CCA)及颈内动脉(ICA)血流,在结扎线近端距分叉 5mm处剪一约 0.3mm小口,将前端加热成圆形、直径为 0.235mm的尼龙线从 ECA残端插入,经 CCA分叉部进入 ICA,向上深入至分叉以上19~20mm,结扎 ECA近心端,10min后抽出栓线,形成再灌注,将线尾埋在皮下,缝和皮肤切口,完成缺血预处理。再灌注 3d后用相同方法再次造成MCAO 2h,第 2次再灌注 22h后处死。以动物清醒后出现同侧 Horner征及对侧前肢为重的瘫痪作为造模成功标准。假手术仅暴露颈总动脉及分叉处,不插线阻断大脑中动脉。各组动物术中均用加热板使其肛温维持在 37℃左右,术后单笼饲养。
1.3 免疫组化染色 每组随机取 6只大鼠,分别于规定时点,以 10%水合氯醛深度麻醉,经左心室灌注生理盐水后,快速断头取前脑,4%中性多聚甲醛固定,脱水,常规石蜡包埋并制成 5μm的切片,每只动物共选取 3张切片,脱蜡至水,梯度酒精脱水,3%H2O2封闭内源性过氧化氢酶、羊血清处理后,加入抗 NF-кB/P65的单克隆抗体 (美国 Santa Cruz公司,1∶50)4℃过夜,依次滴加生物素化羊抗小鼠血清及 SP(工作浓度 1∶200),37℃各孵育60min,DAB显色,苏木素复染,梯度酒精脱水,二甲苯透明,封片。在每张切片随机选取 10个高倍视野进行图像分析,各组所选部位相同。以 PBS代替一抗作为空白对照。
1.4 图像分析 用美国 Nikon Spot图像采集处理系统采图后,采用 Image-Pro Plus 4.1专业图像分析软件进行图像分析,测量免疫组化染色的积分光密度值(IOD)。
1.5 总 RNA提取 每组各取 6只大鼠,处死后立即快速断头,取缺血侧脑组织置于 -80℃冰箱保存待用。采用异硫氰酸胍一步法提取总 RNA,每100mg脑组织加入 1ml Trizol试剂 (美国 Invitrogen公司)。引物按 Genebank所提供的基因序列设计,由 Genebase公司合成。内参照 GAPDH。
1.6 RT-PCR 采用 cDNA逆转录与 PCR扩増一步法试剂盒(Takara公司),在 MJ PTC-200型 PCR仪(美国)中进行 RT-PCR反应。反应液配制:5μl 10×RT-PCR buffer,10μl Mgcl2,5μl dNTP,1μl RNA酶抑制剂,1μl AMV优化的逆转录酶,1μl AMV优化的 Taq DNA聚合酶,RNA及上、下游引物各 1μl,加去除 RNA酶的 H2O至 50μl。反应条件:50℃逆转录 30min,94℃ 2min使逆转录酶失活,94℃变性30s,56℃退火 30s,72℃延伸 1min,扩増 40循环,全部循环结束后于 72℃延伸 10min。取 RT-PCR产物1μl进行 2%琼脂糖凝胶电泳,电压 5V/cm,5μl/ml EB染色,紫外灯下观察、照相,采用 Typhoon 8600多功能激光扫描成像系统(美国)进行图像分析,以看家基因 GAPDH为内参照,COX2 mRNA水平以 PCR产物的光密度与 GAPDH的光密度之比表示。
2 结 果
2.1 NF-ΚB表达变化 3组均有 NF-кB表达,但对照组仅有极少数阳性细胞,其余两组梗死灶周围的半暗区内可见较多的 NF-кB阳性细胞,光镜下胞核呈黄褐色,苏木素不能复染,另有部分细胞为胞浆表达,图像分析示预缺血组 NF-кB表达明显弱于缺血组(P<0.01,见表1、见图1~图3)。
2.2 COX-2mRNA转录 COX-2的 RT-PCR产物 260p,GAPDH产物 600bp,同样反应条件下,各组样本 GAPDH表达基本相同而 COX-2明显不同,对照组仅有极少量表达,预缺血组 COX-2表达明显弱于缺血组(P<0.01,见表1)。
表1 各组 NF-кB表达与 COX-2mRNA水平变化 ±s)

表1 各组 NF-кB表达与 COX-2mRNA水平变化 ±s)
与对照组相比*P<0.01;与缺血组相比#P<0.01
SS+SS IPC+MCAO SS+MCAO 760.92±53.4516098.18±1265.33*23565.81±1978.36*#7.21±0.1939.77±5.01*#92.79±7.62*
3 讨 论
NF-кB是一种多向性转录因子,其结合活性由IкB家族的一组抑制蛋白控制,在静息状态下,NF-кB二聚体与 IкB结合成三聚体,以无活性的形式存在于细胞浆内。当细胞受到缺血等刺激时,IкB激酶活化使 IкB磷酸化并与 NF-кB解聚,从而激活NF-кB。活化的 NF-кB可通过 NLS进入核内进一步调控包括 COX-2在内的多种靶基因转录,在中枢神经系统的信号转导中具有重要意义[4,5]。
近年来研究发现,NF-кB是脑缺血后炎症级联反应的始动因素,可能参与了缺血性脑损伤的病理过程。在 NF-кB的众多靶基因中,COX-2在缺血后信号瀑布中扮演了重要角色[6]。COX-2是花生四烯酸转化为前列腺素的限速酶,在大多数细胞生理条件下不表达,在炎症组织中,可被内毒素和细胞因子诱导而迅速表达,是炎症反应链中的关键因子。与脑缺血后继发的炎症反应与组织损伤密切相关。本研究发现,与对照组相比,给予 2h MCAO的两组动物 NF-кB及COX-2表达均明显增高,提示局灶性脑缺血可能通过激活 NF-кB/COX-2系统,介导了缺血再灌注损伤[7]。KelleyK等发现,基因敲除 COX-2的大鼠,脑缺血后出现 COX-2mRNA不表达和 PGE2表达下降,而且脑损伤体积明显缩小[8],亦支持这一结论。
COX-2介导缺血性脑损伤的机制尚不清楚,COX-2可能涉及了谷氨酸的神经毒性、炎症反应等缺血后早期与迟发的多个病理生理环节,其中,前者是缺血性神经损伤的启动环节,而后者则通过级联反应加剧了继发损伤[9]。COX-2作为前列腺素合成酶,催化 PGG2转变为 PGH2时生成氧自由基,后者能直接导致缺血性神经元坏死,也可通过 NO通路调节氧化应激反应,与 NO形成氧化能力更强的过氧亚硝基,加剧脑损伤。此外,COX-2的过度表达还可改变细胞某些增殖和凋亡相关基因的表达。
尽管已有越来越多的资料证明,NF-кB及其靶基因 COX-2的活化是脑缺血后炎症反应中的重要环节,但目前对其在诱导脑缺血耐受中的意义尚了解甚少。本研究发现,如提前给予缺血预处理,则该组 NF-кB及 COX-2的表达水平明显低于假手术组,提示缺血预处理可能阻断了 IкB激酶的活化,通过减少 IкB的磷酸化,抑制了再次缺血时 NF-кB的激活,进而下调COX-2的基因转录,最终减轻 NF-кB/COX-2介导的缺血后炎症反应,减少了神经元的死亡。缺血预处理抑制 NF-B及COX-2表达的分子机制还未阐明,可能与上调磷脂酰肌醇 3-激酶(PI3-K/Akt)等有关[10],但具体信号转导途径尚有待进一步研究。
综上所述,缺血预处理所诱导的 NF-кB活化减少和 COX-2基因转录下调,可能是脑缺血耐受产生的重要分子机制之一,抑制 NF-кB/COX-2基因转录系统可望增强缺血耐受,从而为寻找缺血性卒中神经保护策略开辟某些新的思路。

图1 SS+SS组 NF-кB表达(免疫组化染色,×400)
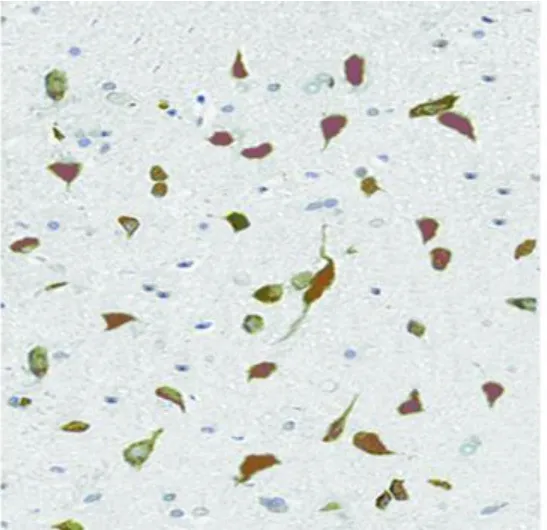
图2 IPC+MCAO组 NF-кB表达 (免疫组化染色,×400)

图3 SS+MCAO组 NF-кB表达 (免疫组化染色,×400)
[1] Hinz B,Brune K.Cyclooxygenase-2~10 years Later[J].J Pharmacol Exp Ther,2002,300:367-375.
[2] Dore S,Otsuka T,Mito T,et al.Neuronal overexpression of cyclooxygenase-2 increases cerebral infarction[J].Ann Neurol,2003,54:155-162.
[3] Yellon DM,Baxter GF,Garcia-Dorado D,et al.Ischemic preconditioning:present position and future directions[J].Cardiovasc Res,1998,37:21-23.
[4] 郝玉曼,罗祖明,周 东.局灶预缺血诱导脑缺血耐受的动物模型[J].中风与神经疾病杂志,2003,20(2):129-131.
[5] Sen R,Baltimore D.Multiple nuclear factor interact with the immunoglobulin enhancer sequences[J].Cell,1986,46(5):705-716.
[6] Lapchak PA,Araujo DM,Song D,et al.Neuroprotection by the Selective Cyclooxygenase-2 Inhibitor SC-236Resultsin Improvements in Behavioral Deficits Induced by Reversible Spinal Cord Ischemia[J].Stroke,2001,32:1220-1225.
[7] Stephenson D,Yin T,Smalstig EB,et al.Transcription factor nuclear factor-kappaB is activated in neurons after focal cerebral ischemia[J].J Cereb Blood Flow Metab,2000,20(3):592-603.
[8] Kelley KA,Ho L,Winger D,et al.Potentiation of excitotoxicity in transgenic mice overexpressing neuronal cyclooxygenase-2[J].Am J Pathol,1999,155:995-1004.
[9] Iadecola C,Niwa K,Nogawa S,et al.Reduced susceptibility to ischemic brain injury and N-methyl-d-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice[J].PNAS,2001,98(3):1294-1299.
[10] Yin W,Signore AP,Iwa M,et al.Preconditioning suppresses inflammation in neonatal hypoxic ischemia via Akt activation[J].Stroke,2007,38:1017-1024.
猜你喜欢
温州大学学报(自然科学版)(2022年2期)2022-05-30
潍坊学院学报(2020年2期)2021-01-18
昆明医科大学学报(2020年11期)2020-12-28
浙江医学(2020年9期)2020-07-01
浙江中西医结合杂志(2019年4期)2019-05-05
浙江医学(2019年2期)2019-01-23
中成药(2018年4期)2018-04-26
制导与引信(2017年3期)2017-11-02
中国继续医学教育(2015年1期)2016-01-06
中国康复理论与实践(2015年10期)2015-12-24
