
基于全基因组关联分析的香蕉枯萎病菌致病基因挖掘与功能研究
2024-11-05 00:00:00高辉蒋尚伯杨迪杜婵娟张晋潘连富崔海涛付岗
南方农业学报 2024年8期









摘要:【目的】深入挖掘香蕉枯萎病菌致病相关基因,并解析其基因特性,为香蕉抗枯萎病品种选育及新药物靶标的开发打下基础。【方法】基于遗传背景将供试299株香蕉枯萎病菌的致病力分别依据数量性状和质量性状进行分组,使用混合线性模型的Gemma软件,运用全基因组关联分析(GWAS)技术对香蕉枯萎病菌致病力表型与全基因组重测序数据进行关联分析;进一步采用ProtParam、SingleIP和SAMRT等生物信息学软件解析候选基因的理化性质及初级结构。【结果】运用GWAS共关联到151个与致病力相关的单核苷酸多态性(SNP)位点。初步确定3个不同类型基因FocScp、FocChp和FocGhf12与枯萎病菌致病力密切相关,进一步对FocScp、FocChp和FocGhf12基因的编码蛋白进行生物信息学分析并预测其功能,结果显示,FocScp基因的cDNA编码区全长948 bp,编码314个氨基酸残基,大小约33.27 kD,含有N-端信号肽,预测为胞外分泌蛋白;FocChp基因的cDNA编码区全长1302 bp,编码433个氨基酸残基,大小约49.17 kD,亚细胞定位在细胞核,预测为含有3个锌指结构域的转录因子;FocGf12基因的cDNA编码区全长1533 bp,编码480个氨基酸残基,大小约53.54 kD,该蛋白存在跨膜结构域,具有GPI修饰位点,亚细胞定位在胞外,预测为糖苷水解酶家族12蛋白。【结论】通过GWAS获得香蕉枯萎病菌3个致病相关基因FocScp、FocChp和FocGhf1 其编码蛋白分别为分泌蛋白、转录因子和结构蛋白。
关键词:香蕉枯萎病菌;致病相关基因;全基因组关联分析;生物信息学分析
中图分类号:S436.681文献标志码:A文章编号:2095-1191(2024)08-2442-12
Mining and functional study of pathogenic genes ofFusariumoxysporumf.sp.cubense based on genome-wideassociation analysis
GAO Hui JIANG Shang-bo YANG Di DU Chan-juan ZHANG Jin PAN Lian-fu CUI Hai-tao 3*,FU Gang1*
(1Plant Protection Research Institute,Guangxi Academy of Agricultural Sciences/Key Laboratory of Green Preventionand Control on Fruits and Vegetables in South China,Ministry of Agriculture and Rural Affairs/Guangxi Key Laboratoryfor Biology of Crop Diseases and Insect Pests,Nanning,GuanZegeB43CpEPO8hsfJeqzd3GZrslJxcw2n9xb8lT+sE4=gxi 530007,China;2College of Agriculture,FujianAgriculture and Forestry University,Fuzhou,Fujian 35300 China;3School of Plant Protection,ShandongAgricultural University,Tai’an,Shandong 272018,China)
Abstract:【Objective】In-depth exploration of the pathogenicity related genes in Fusarium oxysporumf.sp.cubense,and analysis of their genetic characteristics were conducted to laid foundation for the breeding of banana varieties resistant tofusarium wilt,as well as the development of new drug targets.【Method】Based on the genetic backgrounds,the patho-genicity of 299 tested strains ofF.oxysporumf.sp.cubense was grouped accrording to quantity traits and quality traits.Using the Gemma software,which employed a mixed linear model,a genome-wide association study(GWAS)was carried out to perform an association analysis between the pathogenic phenotype ofF.oxysporumf.sp.cubense and the genome-wide resequencing data.Additionally,ProtParam,SingleIP and SAMRT online software tools were utilized to analyze the physicochemical properties and primary structure of candidate genes.【Result】Using GWAS,a total of 151 pathogenicity related single nucleotide polymorphism(SNP)loci were associated.It was preliminarily determined that 3 different types of genes FocScp,FocChp and FocGhf12 were closely related to the pathogenicity ofF.oxysporumf.sp.cubense.Further bioinformatics analysis was performed on the encoded proteins ofFocScp,FocChp and FocGhf12 genes and their func-tions were predicted.The results revealed that the full-length cDNA coding region ofFocScp gene was 948 bp,and en-coded 314 amino acid residues with size of about 33.27 kD and contained an N-terminal signal peptide,which was predicted to be an extracellular secretory protein.The full-length cDNA coding region of FocChp gene was 1302 bp,en-coding 433 amino acid residues with size of about 49.17 kD,with subcellular localization in the nucleus,and was pre-dicted to be a transcription factor containing 3 zinc finger domains.The full-length cDNA coding region of FocGhf12 gene was 1533 bp,encoding 480 amino acid residues with size of about 53.54 kD.The protein had transmembrane domain,GPI modification sites,and subcellular localization outside the cell.It was predicted to be a glycoside hydrolase family 12 protein.【Conclusion】Three pathogenicity related genes(FocScp,FocChp and FocGhf12)ofF.oxysporumf.sp.cubense are obtained by GWAS,and their encodesd proteins are secretory protein,transcription factor and structural protein.
Key words:Fusariumoxysporumf.sp.cubense;pathogenicity related genes;genome-wide association study(GWAS);bioinformatics analysis
Foundation items:National Natural Science Foundation of China(31960520);Central Guidance for Local Science and Technology Development Fund(Guike ZY21195015);Science and Technology Development Project of Guangxi Academy of Agricultural Sciences(2020ZX10,2024YP013)
0引言
【研究意义】由尖孢镰刀菌古巴专化型(Fusarium oxysporumf.sp.cubense,Foc)引起的香蕉枯萎病是香蕉生产中危害最严重的一种土传病害(Ploetz,2006a;Pegg etal.,2019;Yang et al.,2023)。该病最早于1874年在澳大利亚出现(Swarupa et al.,2014;Siamak and Zhang,2018),1967年在我国台湾首次发现,并鉴定出致病菌新的生理小种4号小种(Focrace 4,Foc4),使香蕉产业再次面临严重威胁(Hwang and Ko,2004;Ploetz,2005,2006b)。20世纪90年代,依据遗传背景和致病条件的差异,4号生理小种又进一步区分为热带4号小种(FocTR4)和亚热带4号小种(FocSTR4)(Zhu et al.,2023)。截至目前,Foc共鉴定出4个生理小种:1号生理小种(Foc1)、2号生理小种(Foc2)、3号生理小种(Foc3)和4号生理小种(Foc4),其中,对香蕉产业造成严重危害的为Foc1和Foc4。目前,香蕉枯萎病已遍布全球主要香蕉产区,成为制约香蕉产业发展的关键因素(李华平等,2019)。当前对香蕉枯萎病病原菌致病机制的研究相对滞后,已报道的致病基因多数源自镰刀菌其他专化型的同源基因。对新致病基因的挖掘,可为香蕉枯萎病防治药物的研发提供新靶标,也有助于进一步阐明香蕉枯萎病病原菌的致病机制。【前人研究进展】近年来,随着高通量测序和生物信息学技术的发展,大量物种的基因组已被测序和注释。香蕉枯萎病菌的全基因组测序最早于2014年完成,Guo等(2014)采用第二代测序(NGS)技术完成了1株Foc1菌株(N2)和2株Foc4菌株(B2和II5)的基因组组装。随后,Yun等(2019)使用单分子实时测序(SMRT)完成了Foc1菌株和FocTR4菌株更为完整的基因组组装。全基因组关联分析(GWAS)在动植物功能基因挖掘中具有巨大优势,其通过以大量的单核苷酸多态性(SNP)作为分子遗传标记,对许多自然个体组成的大群体进行基因组水平上的相关分析,从而确定表型性状和遗传位点的相关性(Mohammadi et al.,2020;王艳茹,2022)。张俊(2013)对30份香蕉群体的抗1号和4号枯萎病性状与SSR分子标记进行关联,共计关联到358个变异位点,其中有21个位点与性状相关。Kawicha等(2023)对340份尖孢镰刀菌番茄专化型侵染番茄的致病力表型进行评价,并通过对基因型过滤最终筛选到4478个SNP,随后运用混合线性模型,结合病情严重程度指数(DSI)与SNP进行全基因组关联分析,共计关联到24个与DSI相关的SNP位点。使用Blast N(https://solgenomics.net/tools/blast/)在番茄基因组染色体中搜索基因功能,鉴定到与Fol侵染应答相关的候选基因,包括溶质载体35家族蛋白、锌指蛋白、肽基脯氨酸顺反异构酶样蛋白、富含亮氨酸的重复样蛋白、WW结构域结合蛋白和bZIP型转录因子等。但该方法在真菌中的应用起步较晚,Skelly等(2013)首次将表型数据整合到酿酒酵母全基因组关联分析研究中,测量了22个遗传多样性菌株中的转录结构、蛋白质、代谢物和形态特征,结合酵母菌株的重测序数据展开遗传标记与性状间的关联分析,最终关联到与转录结构和蛋白质丰度显著相关的SNP位点分别有302和64个。Talas等(2016)对220株禾谷镰刀菌分离株进行了与限制性位点相关的DNA测序(RAD-seq),质控后约获得29000个单核苷酸多态性标记,结合菌株侵染性、DON毒素产生及丙环唑敏感性等表型进行全基因组关联分析,共计关联到与侵染性、DON毒素产生及丙环唑敏感性显著相关的SNP位点分别有50、29和74个。【本研究切入点】因Foc遗传背景复杂,其致病机制仍未明晰,生产上也缺乏针对香蕉枯萎病的有效防治药剂及理想抗病品种。致病相关基因的挖掘和病原菌致病机制的解析是推动香蕉枯萎病防控研究的关键环节。【拟解决的关键问题】使用混合线性模型,运用Gemma软件,结合主成分分析和亲缘关系矩阵,对香蕉枯萎病菌进行全基因组关联分析,挖掘致病相关基因,通过生物信息学方法分析其理化性质和初级结构,旨在进一步阐明香蕉枯萎病病原菌的致病机制,为香蕉抗枯萎病品种选育及新药物靶标的开发打下基础。
1材料与方法
1.1试验材料
供试香蕉枯萎病菌菌株299株,分别采集自广东、广西、云南、海南、福建等我国香蕉主产区及缅甸、尼泊尔、越南等香蕉产区(表1),其中1号生理小种和4号生理小种分别有183、116株,均保存于广西农业科学院植物保护研究所。
1.2香蕉枯萎病菌致病力表型测定及性状分组
香蕉枯萎病菌在PDA培养基培养7 d后,用无菌水冲洗菌落获得分生孢子,将孢子悬浮液调整浓度至1×106 CFU/mL。使用4~5叶期的香蕉和粉蕉,采用水培法接种,每株苗接种200 mL孢子悬浮液(1×105 CFU/mL),以接种无菌水为对照,每株菌株设3个重复,每重复3株苗。接种2周后进行调查,记录发病等级并计算病情指数。病情指数=∑(各级病株数×各级代表值)/(调查总株数×最高一级代表值)×100。病情分级标准:0级,球茎呈白色,无褐变;1级,球茎褐变面积≤25%;3级,26%<球茎褐变面积≤50%;5级,51%<球茎褐变面积≤75%;7级,球茎褐变面积>76%。
由于香蕉枯萎病菌具有丰富的遗传多样性,为降低表型数据混杂造成的假阳性,按照生理小种进行分组处理:针对生理小种首先使用总群体进行关联分析,进而以Foc1、Foc4、TR4和SR4等群体进行关联分析。首先使用数量性状方式进行表型性状关联,然后使用质量性状方式进行关联,因其无法使用具体数值衡量,故将Foc1的菌株赋值为0、Foc4的菌株赋值为1进行关联分析。分级性状分布类似质量性状,但实际受多基因控制,故需要具体数值进行分组。因此,根据致病力(病情指数)大小进行分级处理后进行关联分析。
1.3香蕉枯萎病菌基因型数据质控
利用VCFtools(Danecek et al.,2011)将299份Foc样本基因组重测序数据进行基因型质量控制,保留最小质量大于50且覆盖深度在3~20的SNP,剔除最小等位基因小于5%且缺失率大于20%的SNP,保留含有2个ALT变异的位点。
1.4致病力表型与全基因组关联分析
使用Plink(Purcell et al.,2007)将基因型文件转换为二进制格式;利用GCTA(Zhou and Stephens,2012)计算亲缘关系矩阵(G矩阵),用以校正遗传背景,减少假阳性(VanRaden,2008)。同时,使用主成分分析(PCA)作为关联分析的协变量;使用Gemma的混合线性模型[Mixed-linear model,MLM,Y=Wα+Xβ+υ+e模型(即表型=平均数+基因型效应+群体结构+环境)]进行本次全基因组关联分析(马雅杰等,2023);Bonferroni作为全基因组关联分析中简单有效的校正方法,可通过对P值的阈值进行校正来实现消除假阳性结果。根据Bonferroni校正P=1/309747=3.22E-06[即-log10(P)=5.49],以此作为显著性阈值进行筛选(李博等,2013;马雅杰等,2023)。利用R包的CM plot进行关联分析结果的可视化,绘制曼哈顿图及QQ-plot图。
1.5致病相关候选基因功能注释
使用TBtools获得候选基因核苷酸和氨基酸序列,运用eggNOG-mapper(http://eggnog-mapper.embl.de/)进行基因功能注释,通过生物信息平台(https://www.omicstudio.cn/home)制作GO功能注释分析及可视化结果。使用KOBAS v3.0(http://bioinfo.org/kobas)进行KEGG信号通路富集分析及可视化结果。
1.6关键致病基因筛选与功能预测
使用TBtools通过比对Foc4基因组数据获得显著位点的核苷酸和氨基酸序列,利用Uniprot(https://www.uniprot.org/)蛋白数据库进行比对,初步确定蛋白功能信息;利用NCBI-BLAST在线软件进行同源性分析,结合二者结果进行文献检索。
使用ProtParam(https://www.expasy.org/resources/protparam)初步分析基因编码蛋白的理化性质;利用ProtScale(https://web.expasy.org/protscale/)和NetPhos(https://services.healthtech.dtu.dk/services/NetPhos-3.1/)分别预测蛋白亲疏水性和磷酸化位点;使用SingleIP 5.0(https://services.healthtech.dtu.dk/ser-vices/SignalP-5.0/#main-content)预测编码蛋白是否存在信号肽位点;使用DTU/DeepTMHMM(https://dtu.biolib.com/DeepTMHMM)预测基因编码蛋白的跨膜结构域;利用Euk-mPLoc 2.0 server(http://www.csbio.sjtu.edu.cn/bioinf/euk-multi-2/#)预测蛋白亚细胞定位;利用big-PI-predictor(https://mendel.imp.ac.at/gpi/fungi_server.html)预测蛋白GIP修饰位点;利用SAMRT(https://smart.embl.de/)和CD-Search(https://www.ncbi.nlm.nih.gov/cdd/)预测蛋白功能结构域。
1.7总RNA提取及反转录
采用TRIzol法提取香蕉枯萎病菌4号生理小种Foc1594菌株的总RNA。称取0.5 g菌丝,用液氮磨成粉末,加入1 mL TRIzol试剂,充分混匀,室温放置5 min;加入200μL氯仿,剧烈震动15 s后室温放置3 min;4℃下12000 r/min离心15 min;取上清400μL至1.5 mL RNA-Free离心管,加入等体积异丙醇颠倒混匀,室温静置2~5 min;4℃下12000 r/min离心10 min;弃上清,加入1 mL 75%乙醇颠倒混匀;4℃下7500 r/min离心5 min后,弃上清,开盖放入烘箱中烘10min;加入30~50μL DEPC水溶解,闭盖放入65℃烘箱中烘10min。将经检测完整的RNA参照TaKaRa逆转录试剂盒操作方法反转录合成cDNA(魏巍等,2012)。
1.8关键致病基因的PCR扩增
通过比对参考基因组获得3个基因的相关信息,分别设计FocScp、FocChp和FocGhf12基因引物Scp-F(5'-ATGCGTTCTCTTCCCATTGCA-3')和Scp-R(5'-CTAAGGACCACCAGCGCG-3')、Chp-F(5'-ATG AGCGCGTCAGGCTCG-3')和Chp-R(5'-CTAGAAC CCAATGAATGCATTCTT-3')、Ghf12-F(5'-ATGGGT AACGCTTCATCGAAAGAT-3')和Ghf12-R(5'-TCA AGAAAGAACCACGCTTATGAC-3')对4号生理小种的基因组DNA进行PCR扩增。反应体系50μL:TaKaRa Mix 25μL,上、下游引物各1μL,DNA模板1μL,ddH2O补足至50μL。扩增程序:98℃预变性5 min;98℃30 s,58℃30 s,72℃1 min,进行35个循环;72℃延伸10 min。
2结果与分析
2.1香蕉枯萎病菌致病力表型及分布
本研究关联使用的299株菌株包含1号生理小种183株、4号生理小种116株,所有菌株均采用水培法完成致病力表型测定(图1)。其中,侵染香蕉苗测得致病力分布情况为0~10占群体的33.4%,10~20占18.7%,20~30占9.4%,30~40占8.4%,40~50占7.4%,50~60占6.4%,60~70占3.7%,70~80占5.0%,80~90占4.0%,90~100占3.6%;侵染粉蕉苗测得致病力分布情况为0~10占群体的4.3%,10~20占7.7%,20~30占5.4%,30~40占5.4%,40~50占10.0%,50~60占8.4%,60~70占12.4%,70~80占11.0%,80~90占15.4%,90~100占20.0%。
考虑到小种间的差异,分别使用Foc1和Foc4的致病力以数量性状方式作为表型数据,根据统计学分析(表2)和直方图(图2),Foc1侵染香蕉致病力的平均数为16.746,峰度和偏度分别为6.656、2.556,且平均数大于中位数,分布为右偏(正偏态),而侵染粉蕉致病力的平均数为72.35 峰度和偏度分别为0.304、-1.030,且平均数小于中位数,分布左偏(负偏态);Foc4侵染香蕉致病力平均数为49.766,且平均数约等于中位数,数值相对集中,峰度和偏度分别为-0.555、0.047;侵染粉蕉致病力平均数为45.866,与中位数接近,峰度和偏度分别为-0.909、0.095。
2.2全基因组关联分析定位致病相关位点
基于筛选获得的309747个高密度SNP和299份香蕉枯萎病菌致病力表型,综合考虑群体结构,利用Gemma软件中混合线性模型结合不同分组方式的致病力进行全基因组关联分析,共关联到151个与致病力相关的SNP位点。其中以Foc4群体作为质量性状,共关联到123个相关SNP位点(图3-A);以Foc4作为数量性状,在2号染色体上关联到3个相关SNP位点(图3-B)。可能受表型数据的影响,将生理小种分组后以Foc1群体数量性状关联未筛选到相关位点;以质量性状关联筛选到几处,但不符合影响表型性状的连锁不平衡状态(图3-C)。根据致病力大小进行分级处理后,仅在以香蕉为寄主的菌株致病力80~100群体中分别在9号及11号染色体中关联到17和8个相关SNP位点(图3-D)。
2.3候选基因功能注释
利用eggNOG-mapper对候选基因进行GO功能注释分析(图4-A),在GO数据库中根据功能分为生物过程(Biological process,BP)、细胞组分(Cellular component,CC)和分子功能(Molecular function,MF),在生物过程富集到25个二级分类,主要在有机物与离子的运输、有机物的代谢过程、应激反应及辅助定位等方面发挥功能;在细胞组成富集到15个二级分类,主要在细胞器及质膜等部位发挥作用;在分子功能富集到10个二级分类,具有一些跨膜转运体活性、有机化合物结合及ATP酶活性相关功能。
使用KOBAS v3.0对候选基因进行KEGG信号通路富集分析(图4-B),发现候选基因主要富集在新陈代谢(Metabolism)、环境信息处理(Environmental information processing)、遗传信息处理(Genetic in-formation processing)和生物体系统(Organismal sys-tems),其中参与代谢途径的基因共有26个,占比68.42%,主要包括代谢途径、脂肪酸生物合成、缬氨酸、亮氨酸和异亮氨酸降解;参与环境信息处理的基因有4个,占比10.52%,其中包括ABC转运途径和AMPK信号通路;参与遗传信息处理的基因有5个,占比13.16%,主要包括抗叶酸抗性通路、谷胱甘肽代谢和碳代谢;参与生物体系统的基因有3个,占比7.89%,主要包括胆汁分泌通路和胰岛素信号通路。
2.4关键致病基因功能解析
通过检索Uniprot蛋白数据库,查询候选基因的遗传背景,结合全基因组关联分析关联的P值和候选基因的注释结果,筛选出3个与致病力密切相关的基因,其编码蛋白分别为FocScp、FocChp和Foc-Ghf12。
FocScp基因全长1134 bp,cDNA编码区全长948 bp,编码314个氨基酸残基,大小约33.27 kD,理论等电点(pI)5.5 半胱氨酸含量3.8%。预测Foc-Scp编码蛋白在N端第1~17位氨基酸位点存在信号肽片段,亚细胞定位在胞外,无跨膜结构域和GPI修饰位点,存在多个磷酸化位点,具有亲水性,预测为分泌蛋白(图5)。
FocChp基因全长2848 bp,cDNA编码区全长1302 bp,编码433个氨基酸残基,大小约49.17 kD,理论等电点(PI)5.78,半胱氨酸含量2.5%。FocChp蛋白不存在N-端信号肽、GPI修饰位点和跨膜结构域,亚细胞定位在细胞核,存在多个磷酸化位点,具有亲水性,预测为含有3个锌指结构域的转录因子(图6)。
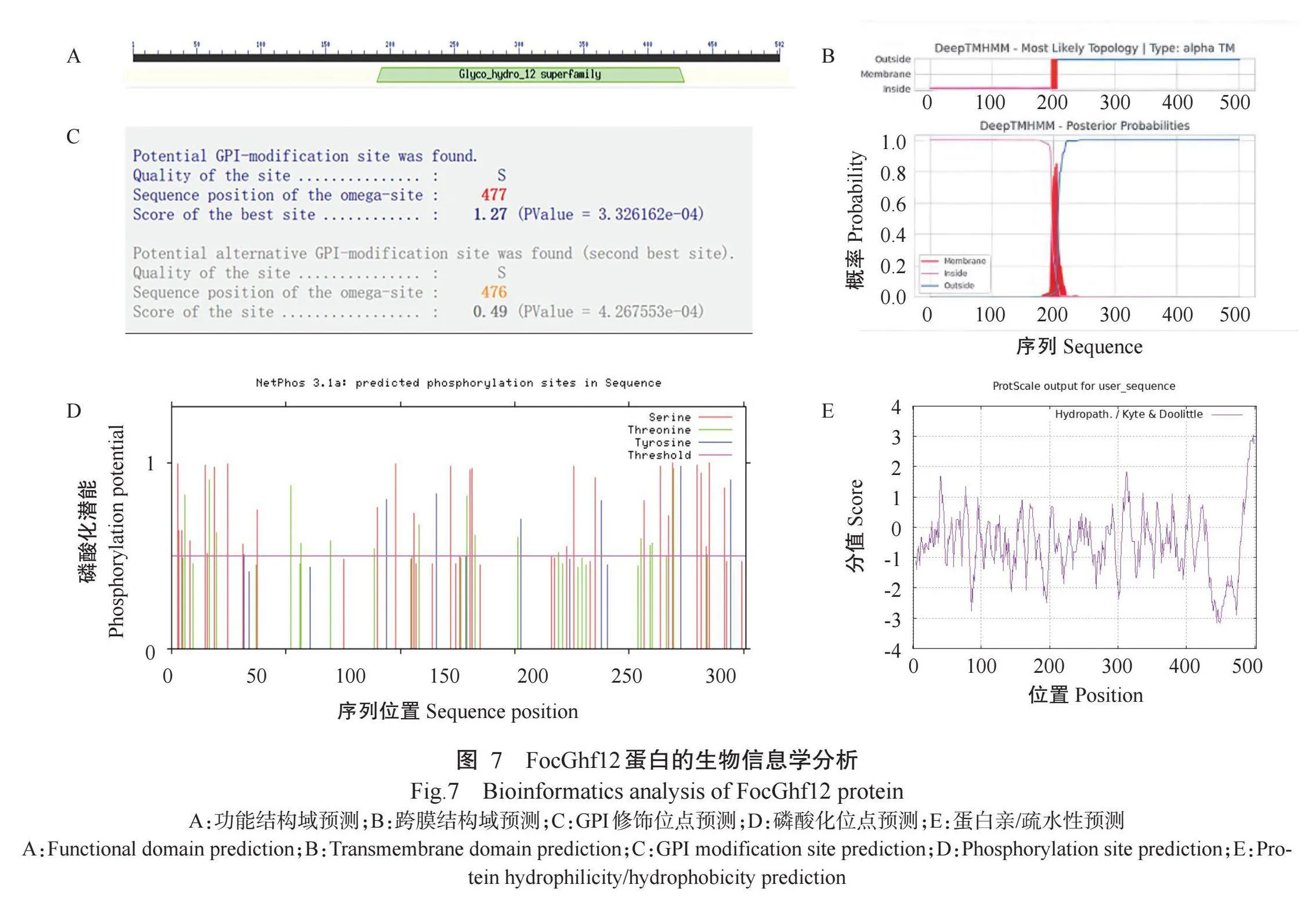
FocGhf12基因全长2351 bp,cDNA编码区全长1533 bp,编码480个氨基酸残基,大小约53.54 kD,理论等电点(pI)5.78,其中半胱氨酸含量2.5%。跨膜结构域预测FocGhf12蛋白结果显示存在可能性达到0.8,不存在N-端信号肽,亚细胞定位在胞外,在466与467两个位点存在GPI修饰位点,存在多个磷酸化位点,为亲水蛋白,预测为糖苷水解酶家族12蛋白(图7)。
2.5关键致病基因克隆验证
提取香蕉枯萎病菌4号生理小种Foc1594菌株的总RNA,以反转录后的cDNA为模板,分别使用Scp-F/R、Chp-F/R和Ghf12-F/R 3对引物进行PCR扩增,分别可以扩增到945、1302和1509 bp大小的条带,经过测序比对,与预测片段大小一致(图8)。
3讨论
随着高通量测序技术的出现,更为全面、深刻和精细地解析复杂性状形成的分子机制,结合转录组学、蛋白质组学和代谢组学等数据进行多组学整合分析也逐渐成为一种趋势(王博和孙广宇,2016;陈元军等,2023)。本研究利用全基因组关联分析技术对299株来自我国广东、广西、云南、海南、福建等香蕉主产区及缅甸、尼泊尔、越南等地的枯萎病菌致病表型性状与全基因组数据进行关联分析,确定了与枯萎病菌致病力密切相关的3个不同类型基因。前人运用全基因组关联分析技术主要是针对植物挖掘抗病基因,而针对病原菌全基因组关联分析的报道较少。Dalman等(2013)首次把全基因组关联分析应用到植物病原真菌中,分析了一种坏死性病原体Heterobasidionannosum对其寄主欧洲云杉(Picea abies)和欧洲赤松(Pinus sylvestris)的毒力;对23个单倍体H.annosum分离株的基因组进行了测序,以菌株对寄主的毒力为表型,通过全基因组关联分析关联到12个与毒力相关的SNP位点。
全基因组关联分析技术通过在整个基因组范围内对SNPs的扫描标记,来定位与特定生物学性状相关的遗传变异。目前在真菌中开展的全基因组关联分析研究多使用单性状和SNP的关联分析,但很多性状同时受多基因控制,倘若单个个体有多个测量性状时,性状间也可能存在关联,这些因素往往被忽视,为降低误差,应在关联时将该因素考虑在内,从而提高检测效率。无论是多性状还是单性状关联,基因型数据与表型数据是直接影响全基因组关联分析结果的2个关键数据。精确的表型检测是关联分析的关键之一,表型数据需根据群体物种生物学特性和遗传特性尝试不同分组,权衡更适宜的方案。对于表型数据,一般采用数量性状、质量性状和分组性状的处理方式。数量性状在自然界中是一个重要特征(Mareesetal.,2018;曹磊等,2023),具有数据分布连续、受环境影响大等特点。但与致病性相关的数量性状在真菌生物学中很少受到关注(Pariaud et al.,2009;Lannou,2012),数量性状受多基因控制,需测量到具体数值,而关联分析属于线性模型,数据必须符合正态分布,需去除极端异常值,防止由于异常值的存在导致假阳性关联偏高。质量性状受单基因控制,无法使用具体数值衡量,因此每个群体选取近似的样本即可。分级性状作为一种处理方式,其表型分布与质量性状类似,但实际上受多基因控制。本研究对3种性状的表型处理均有尝试。首先使用具体的香蕉或粉蕉致病力数值进行关联,但表型数据不符合正态分布,故未关联到理想结果,继而根据菌株遗传特性剔除离群数据,但效果也不理想,最终以Foc4为群体进行关联,在2号染色体上关联到3个显著SNP位点;随后尝试质量性状,根据生理小种进行赋值,其中以Foc4作为质量性状,共关联到123个相关SNP位点;最后使用分级性状,根据不同致病力划分等级,但只在香蕉为寄主的菌株致病力分级线设置为80时,以此群体数据分别在9号及11号染色体中关联到17和8个相关SNP位点。据此共计关联到151个与Foc致病相关的SNP位点。对于该群体规模,仅关联到151个SNP位点,结果少于预期,但为香蕉枯萎病菌在基因组层面的分析打下了基础。后续将对该群体的表型进行深入探究,结合其他表型数据重新进行关联。
本研究根据关联到的SNP位点,检索到香蕉枯萎病菌的大量致病相关候选基因;并通过比对Uni-prot蛋白数据库,发现这些基因所编码的蛋白包括锌蔟家族蛋白、CFEM(Common in fungal extracellu-lar membrane)结构域蛋白、含有P环的核苷三磷酸水解酶超家族蛋白、富含半胱氨酸的小分泌蛋白、C2H2型转录因子及糖苷水解酶12家族蛋白等。其中有些蛋白在植物病原真菌中已有报道,如Zhang等(2023)在假禾谷镰刀菌中也鉴定到锌蔟家族转录因子FpUme18,靶向敲除后发现∆Fpume18缺失突变体在生长、分生孢子产生和分生孢子萌发方面存在缺陷,同时降低了小麦胚芽鞘的致病性。此外,FpUme18也通过调节TRI基因表达参与DON毒素的产生,表明FpUme18参与调控假禾谷镰刀菌的毒力。CFEM是真菌中特有的一类位于细胞外膜的蛋白,许多CFEM结构蛋白被证明在病原真菌与寄主植物互作中起效应因子的作用(Kulkarni et al.,2003;Zhang et al.,2015)。Huang等(2023)从甘蔗镰刀菌基因组中鉴定到20个CFEM蛋白,其中4个FsC-FEM蛋白(Fs06761、Fs08184、Fs10706和Fs13617)能抑制烟草上Bax(BCL2-associated X protein)所诱导的细胞程序性死亡;使用YTK12酵母系统验证其为分泌蛋白,靶向敲除后,Fs06761、Fs08184和Fs13617缺失造成甘蔗镰刀菌致病力降低,表明3个基因是病原菌的重要致病因子。同时,发现Scp同源蛋白在尖孢镰刀菌和亚洲大豆锈病中曾有报道。Li等(2019)在分泌组中发现Cerato-platanin(CP)蛋白家族的一个成员FocCP 该蛋白可以被植物的病原体相关分子模式(PAMP)所识别,并引起一系列免疫反应,此外还能增强烟草对烟草花叶病毒及香蕉对Foc4的抗性。Chp在番茄枯丝核菌和梨树腐烂病菌曾有报道,Ghosh等(2021)通过HIGS介导的基因沉默将转录组中一些上调基因沉默后发现,RS_CRZ1(C2H2型锌指转录因子)沉默植株不仅表现出疾病症状的显著减少,而且病原体的定殖也受到损害。Ghf12在棉花枯萎病及番茄枯萎病中曾有报道,Zhang等(2021)发现一种由尖孢镰刀菌分泌的糖苷水解酶家族12蛋白FoEG 并证明该蛋白可触发不同植物的细胞死亡,诱导植物的防御反应,当FoEG1缺失或酶活性丧失时会降低尖孢镰刀菌的毒力。可以看出,利用全基因组关联分析技术挖掘植物病原菌致病基因是较为可靠并高效的方法。
4结论
本研究以299份香蕉枯萎病菌作为关联群体,结合309747个高密度SNP,针对Foc对香蕉和粉蕉致病力性状进行全基因组关联分析,共关联到151个与致病力相关的SNP位点,根据基因功能注释及功能预测,确定了3个与枯萎病菌致病力密切相关的不同类型基因FocScp、FocChp和FocGhf1 其编码蛋白分别为分泌蛋白、转录因子和结构蛋白。
参考文献(References):
曹磊,毛文文,梁晓雪,李翔,王盼乔,侯娟,李琼,胡建斌.2023.甜瓜叶绿素含量全基因组关联分析及候选基因预测[J].河南农业大学学报,57(2):231-240.[Cao Lei,Mao W W,Liang X X,Li X,Wang P Q,Hou J,Li Q,Hu J B.2023.Genome-wide association analysis of chlorophyll content in melon and prediction of the candidate genes[J].Journal of Henan Agricultural University,57(2):231-240.]doi:10.16445/j.cnki.1000-2340.20230224.002.
陈元军,马娟娟,史睿,李伟龙,王婷,彭琦,张维,陈锋,王晓东,高建芹,付三雄,张洁夫,孙程明,季彪俊,胡茂龙.2023.整合关联分析和共表达网络分析挖掘甘蓝型油菜籽粒质量候选基因[J].江苏农业学报,39(4):913-930.[Chen Y J,Ma J J,Shi R,Li W L,Wang T,Peng Q,Zhang W,Chen F,Wang X D,Gao J Q,Fu S X,Zhang J F,Sun C M,Ji B J,Hu M L.2023.Integrating genome-wide associa-tion study and weighted gene co-expression network analy-sis to explore candidate genes of seed weight in rapeseed(Brassica napus L.)[J].Jiangsu Journal of Agricultural Sciences,39(4):913-930.]doi:10.3969/j.issn.9fc693c5f1392cce329c763173845ebd1000-4440.2023.04.001.
李博,张焕欣,杨小艳,吕颖颖,江培顺,郝转芳,吕香玲,王宏伟,翁建峰.2013.玉米穗位高全基因组关联分析及其候选基因预测[J].作物杂志,(2):27-32.[Li B,Zhang H X,Yang X Y,LüY Y,Jiang P S,Hao Z F,LüX L,Wang H W,Weng J F.2013.Genome-wide association study and candidate gene prediction of ear height in maize(Zea mays L.)[J].Crops,(2):27-32.]doi:10.16035/j.issn.1001-7283.2013.02.026.
李华平,李云锋,聂燕芳.2019.香蕉枯萎病的发生及防控研究现状[J].华南农业大学学报,40(5):128-136.[Li H P,Li Y F,Nie Y F.2019.Research status of occurrence and control of fusarium wilt of banana[J].Journal of South China Agricultural University,40(5):128-136.]doi:10.7671/j.issn.1001-411X.201905062.
马雅杰,鲍建喜,高悦欣,李雅楠,秦文萱,王彦博,龙艳,李金萍,董振营,万向元.2023.玉米株高和穗位高性状全基因组关联分析[J].作物学报,49(3):647-661.[Ma Y J,Bao J X,Gao Y X,Li Y N,Qin W X,Wang Y B,Long Y,Li J P,Dong Z Y,Wan X Y.2023.Genome-wide associa-tion analysis of plant height and ear height related traits in maize[J].ActaAgronomica Sinica,49(3):647-661.]doi:10.3724/SP.J.1006.2023.23023.
王博,孙广宇.2016.基于高通量测序的群体基因组学——植物病原真菌研究新方向[J].菌物学报,35(12):1434-1440.[Wang B,Sun G Y.2016.Population genomics based on high-throughput sequencing—A new research field in plant pathogenic fungi[J].Mycosystema,35(12):1434-1440.]doi:10.13346/j.mycosystema.160224.
王艳茹.2022.苦瓜种质枯萎病抗性全基因组关联分析[D].海口:海南大学.[Wang Y R.2022.Genome-wide associa-tion analysis of resistance to fusarium wilt in bitter gourd germplasm[D]Haikou:Hainan University.]doi:10.27073/d.cnki.ghadu.2022.000941.
魏巍,杨腊英,周端咏,谢德啸,黄小娟,刘一贤,黄俊生.2012.香蕉枯萎病菌fgb2基因的克隆与序列分析[J].热带生物学报,3(2):138-141.[WeiW,Yang LY,Zhou DY,Xie D X,Huang X J,Liu Y X,Huang J S.2012.Cloning and sequence analysis offgb2 gene in Fusarium oxyspo-rum f.sp.cubense[J].Journal of Tropical Organisms,3(2):138-141.]doi:10.3969/j.issn.1674-7054.2012.02.011.
张俊.2013.香蕉枯萎病抗性的种质筛选及其枯萎病抗性基因的初步关联分析[D].海口:海南大学.[Zhang J.2013.Screening on banana germplasm for resistance to fusarium wilt and the preliminary correlation analysis of its resis-tance genes[D].Haikou:Hainan University.]
Dalman K,Himmelstrand K,OlsonÅ,Lind M,Brandström-Durling M,Stenlid J.2013.A genome-wide association study identifies genomic regions for virulence in the non-model organism Heterobasidionannosums.s[J].PLoS One,8(1):e53525.doi:10.1371/journal.pone.0053525.
Danecek P,Auton A,Abecasis G,Albers C A,Banks E,DePristo M A,Handsaker R E,Lunter G,Marth G T,Sherry S T,McVean G,Durbin R.2011.The variant call format and VCFtools[J].Bioinformatics,27(15):2156-2158.doi:10.1093/bnfrmatcs/btr330.
Guo L J,Han L J,Yang L Y,Zeng H C,Fan D D,Zhu Y B,Feng Y,Wang G F,Peng C F,Jiang X T,Zhou D J,Ni P X,Liang C C,Liu L,Wang J,Mao C,Fang X D,Peng M,Huang J S.2014.Genome and transcriptome analysis of the fungal pathogen Fusarium oxysporumf.sp.cubense causing banana vascular wilt disease[J].PLoS One,9(4):e95543.doi:10.1371/journal.pone.0095543.
Ghosh S,Kant R,Pradhan A,Jha G.2021.RS_CRZ a C2H2-type transcription factor is required for pathogenesis of Rhizoctonia solani AG1-IA in tomato[J].Molecular Plant-Microbe Interactions,34(1):26-38.doi:10.1094/MPMI-05-20-0121-R
Huang Z,Zhou Y M,Li H X,Bao Y X,Duan Z Z,Wang C X,Powell C A,Wang K,Hu Q,Chen B S,Zhang J S,Zhang M Q,Yao W.2023.Identification of common fungal extra-cellular membrane(CFEM)proteins in Fusarium sacchari that inhibit plant immunity and contribute to virulence[J].Microbiology Spectrum,11(6):e0145223.doi:10.1128/spectrum.01452-23.
Hwang S C,Ko W H.2004.Cavendish banana cultivars resis-tant to Fusarium wilt acquired through somaclonal varia-tion in Taiwan[J].Plant Disease,88(6):580-588.doi:10.1094/PDIS.2004.88.6.580.
Kawicha P,Tongyoo P,Wongpakdee S,Rattanapolsan L,Duangjit J,Chunwongse J,Suwor P,Sangdee A,Thanya-siriwat T.2023.Genome-wide association study revealed genetic loci for resistance tofusarium wilt in tomato germ-plasm[J].Crop Breeding and Applied Biotechnology,23(1):e43532311.doi:10.1590/1984-70332023v23n 1a1.
Kulkarni R D,Kelkar H S,Dean R A.2003.An eight-cysteine-containing CFEM domain unique to a group of fungal membrane proteins[J].Trends in Biochemical Sciences,28(3):118-121.doi:10.1016/S0968-0004(03)00025-2.
Lannou C.2012.Variation and selection of quantitative traits in plant pathogens[J].Annual Review of Phytopathology,50:319-338.doi:10.1146/annurev-phyto-081211-173031.
Li S W,Dong Y J,Li L,Zhang Y,Yang X F,Zeng H M,Shi M F,Pei,X W,Qiu D W,Yuan Q H.2019.The novel Cerato-Platanin-Like Protein FocCP1 from Fusarium oxysporum triggers an immune response in plants[J].International Journal of Molecular Sciences,20(11):2849.doi:10.3390/ijms20112849.
Marees A T,De Kluiver H,Stringer S,Vorspan F,Curis E,Marie-Claire C,Derks E M.2018.A tutorial on conducting genome-wide association studies:Quality control and sta-tistical analysis[J].International Journal of Methods in Psy-chiatric Research,27(2):e1608.doi:10.1002/mpr.1608.
Mohammadi M,Xavier A,Beckett T,Beyer S,Chen L Y,Chikssa H,Cross V,Moreira F F,French E,Gaire R,Griebel S,Lopez M A,Prather S,Russell B,Wang W D.2020.Identification,deployment,and transferability of quantitative trait loci from genome-wide association stu-dies in plants[J].Current Plant Biology,24:100145.doi:10.1016/j.cpb.2020.100145.
Pariaud B,RavignéV,Halkett F.2009.Aggressiveness and its role in the adaptation of plant pathogens[J].Plant Patho-logy,58(3):409-424.doi:10.1111/j.1365-3059.2009.020 39.x.
Pegg K G,Coates L M,O’Neill W T,Turner D W.2019.The epidemiology offusarium wilt of banana[J].Frontiers in Plant Science,10:1395.doi:10.3389/fpls.2019.01395.
Ploetz R C.2005.Panama disease:An old nemesis rears its ugly head:Part 1.The beginnings of the banana export trades[J].Plant Health Progress,6(1):18.doi:10.1094/PHP-2005-1221-01-RV.
Ploetz R C.2006a.Fusarium wilt of banana is caused by seve-ral pathogens referred to as Fusarium oxysporumf.sp.cubense[J].Phytopathology,96(6):653-656.doi:10.1094/PHYTO-96-0653.
Ploetz R C.2006b.Panama disease:An old nemesis rears its ugly:Head part 2.The Cavendish era and beyond[J].Plant Health Progress,7(1):36.doi:10.1094/PHP-2006-0308-01-RV.
Purcell S,Neale B,Todd-Brown K,Thomas L,Ferreira M A,Bender D,Maller J,Sklar P,De Bakker P I,Daly M J,Sham P C.2007.
PLINK:A tool set for whole-genome association and population-based linkage analyses[J].Ame-rican Journal of Human Genetics,81(3):559-575.doi:10.1086/519795.
Siamak S B,Zhang S J.2018.Banana fusarium wilt(Fusarium oxysporumf.sp.cubense)control and resistance,in the context of developing wilt-resistant bananas within sustaina-ble production systems[J].Horticultural Plant Journal,4(5):208-218.doi:10.1016/j.hpj.2018.08.001.
Skelly D A,Merrihew G E,Riffle M,Connelly C F,Kerr E O,Johansson M,Jaschob D,Graczyk B,Shulman N J,Wakefield J,Cooper S J,Fields S,Noble W S,Müller E G D,Davis T N,Dunham M J,MacCoss M J,Akey J M.2013.Integrative phenomics reveals insight into the struc-ture of phenotypic diversity in budding yeast[J].Genome Research,23(9):1496-1504.doi:10.1101/gr.155762.
Swarupa V,Ravishankar K V,Rekha A.2014.Plant defense response against Fusarium oxysporum and strategies to develop tolerant genotypes in banana[J].Planta:An Inter-national Journal of Plant Biology,239(4):735-751.doi:10.1007/s00425-013-2024-8.
Talas F,Kalih R,Miedaner T,McDonald B A.2016.Genome-wide association study identifies novel candidate genes for aggressiveness,deoxynivalenol production,and azole sen-sitivity in natural field populations of Fusarium gra-minearum[J].Molecular Plant-Microbe Interactions,29(5):417-430.doi:10.1094/MPMI-09-15-0218-R.
VanRaden P M.2008.Efficient methods to compute genomic predictions[J].Journal of Dairy Science,91(11):4414-4423.doi:10.3168/jds.2007-0980.
Yang Y B,An B,Guo Y F,Luo H L,He C Z,Wang Q N.2023.A novel effector,FSE regulates the pathogenicity of Fusarium oxysporumf.sp.cubense tropical race 4 to banana by targeting the MYB transcription factor MaEFM-Like[J].Journal of Fungi,9(4):472.doi:10.3390/jof90 40472.
Yun Y Z,Song A X,Bao J D,Chen S S,Lu S M,Cheng C Z,Zheng W H,Wang Z H,Zhang L S.2019.Genome data of Fusarium oxysporumf.sp.cubense race 1 and tropical race 4 isolates using long-read sequencing[J].Molecular Plant-Microbe Interactions,32(10):1270-1272.doi:10.1094/MPMI-03-19-0063-A.
Zhang L,Yan J P,Fu Z C,Shi W J,Ninkuu V,Li G Y,Yang XF,Zeng H M.2021.FoEG a secreted glycoside hydro-lase family 12 protein from Fusarium oxysporum,triggerscell death and modulates plant immunity[J].MolecularPlant Pathology,22(5):522-538.doi:10.1111/mpp.13041.
Zhang Y,Zhuang X Y,Meng J X,Zan F F,Liu Z R,Qin C C,Hao L J,Wang Z F,Wang L M,Li H L,Li H Y,Ding S L.2023.A putative Zn(II)2Cys6-Type transcription factorFpUme18 is required for development,conidiation,cellwall integrity,endocytosis and full virulence in Fusariumpseudograminearum[J].International Journal of Molecu-lar Sciences,24(13):10987.doi:10.3390/ijms241310987.
Zhang Z N,Wu Q Y,Zhang G Z,Zhu Y Y,Murphy R W,Liu Z,Zou C G.2015.Systematic analyses reveal uniqueness and origin of the CFEM domain in fungi[J].Scientific Reports,5:13032.doi:10.1038/srep 13032.
Zhou X,Stephens M.2012.Genome-wide efficient mixed-model analysis for association studies[J].Nature Genetics,44(7):821-824.doi:10.1038/ng.2310.
Zhu Z Y,Wu G Y,Deng R F,Hu X Y,Tan H B,Chen Y P,Tian Z H,Li J X.2023.Spatiotemporal biocontrol and rhi-zosphere microbiome analysis of Fusarium wilt of banana[J].Communications Biology,6(1):27.doi:10.1038/s42003-023-04417-w.
(责任编辑麻小燕)
