
不同提取方式对金针菇菇脚蛋白性质的影响
2024-04-29 06:39贾明月贵香茹原秋艳陈美玲董晓博徐怀德
食品工业科技 2024年8期
贾明月,贵香茹,原秋艳,陈美玲,简 磊,董晓博,徐怀德
(西北农林科技大学食品科学与工程学院,陕西咸阳 712100)
金针菇是世界上工业化大规模生产水平最高的食用菌之一,据统计,2020 年金针菇年产量约为227.91 万吨[1]。随着金针菇产量的逐年增加,随之而来的是其副产物金针菇菇脚(Flammulinavelutipesstembase)的大量产生。金针菇菇脚(指菇柄底部及带有培养基部分)水分含量高,并且具有多种营养物质,极易腐败变质。如果处理不当,会造成严重的环境污染[2]。通过对金针菇生产企业调查发现,当前,金针菇菇脚的处理方式主要有两种,一种是卖给肥料公司当动物饲料或堆肥处理,另一种是混合煤炭在锅炉中烧掉。所以按照目前的处理方式,金针菇菇脚并没有被合理利用[3]。金针菇菇脚中含有蛋白质、多糖、纤维素、多酚、黄酮、皂苷等多种生物活性物质,具有免疫活性、抗氧化、抗菌、抗肿瘤等多种生物活性[4-5]。其中,蛋白质作为菇脚的基本营养成分之一,约占其干重的10%~20%[6]。而目前,对于金针菇菇脚的研究主要集中在增加金针菇产量以及用作饲料喂养家禽等方面,而对于金针菇菇脚蛋白的研究较少。林忠宁等[7]测定了金针菇菇脚中的蛋白质以及氨基酸含量,结果表明其氨基酸种类齐全,含有较丰富的谷氨酸和天冬门氨酸,其中人体必需的赖氨酸色氨酸含量分别达到13.02 mg/g、14.68 mg/g,与鸡蛋蛋白的贴近度达0.8257,是一种良好的氨基酸来源。金针菇菇脚进行水解可以生产蛋白小肽,得到的蛋白小肽粉末富含VB1、VB2、VC等多种维生素和钙、磷、铁等多种矿物质,营养丰富,易被人体吸收,具有优异的抗氧化性,可用于制备功能性食品。因此从金针菇菇脚中提取蛋白质具有很大的实用意义。
目前对于植物蛋白提取主要有水提法、碱提法、酶法和有机溶剂提取法等,其中碱提法因提取率高、操作简单而被广泛应用,已经成功运用于各种植物蛋白的提取,各种物理辅助法也有较为广泛的应用[8]。崔晓瑞等[9]优化了超声辅助碱法提取大球盖菇蛋白的工艺,其产物具有一定的热稳定性。Jin 等[10]采用盐析和等电点沉淀两种不同的方法提取平菇蛋白,结果发现等电点沉淀的蛋白具有更高的蛋白含量,盐析法的蛋白具有更高的总酚含量。李妍等[11]用胶体磨辅助酶法提取鹿茸蛋白,结果发现产物具有良好的免疫活性。张艳荣等[12]优化了超声-微波复合辅助碱法提取白灵菇蛋白。刘家希等[13]发现超声辅助碱法能够有效提高米糠蛋白质的提取率和功能性质。许凤等[14]发现胶体磨超声辅助碱法能够显著提高米糠蛋白的提取率。蒋益等[15]优化了冻融辅助碱法从蛹虫草中提取蛋白质的工艺。研究发现,物理辅助碱提会对蛋白性质具有一定影响[14],而目前的研究多集中在物理辅助碱法的优化上。
因此,本实验拟采用碱提法、超声辅助碱提法、胶体磨辅助碱提法、胶体磨联合超声辅助碱提法以及冻融辅助碱提法提取金针菇菇脚中的蛋白质,比较几种提取方法下菇脚蛋白功能性质(持水性、持油性、起泡及起泡稳定性、乳化及乳化稳定性、溶解性)、理化性质(电位、粒径、表面疏水性、巯基)及结构性质(红外光谱、荧光光谱、二级结构)的差异,为金针菇生产的副产物的综合利用提供一定的理论依据。
1 材料与方法
1.1 材料与仪器
新鲜金针菇菇脚(白金针菇)陕西众兴高科食用菌有限公司;牛血清白蛋白、十二烷基硫酸钠、考马斯亮蓝G250、磷酸、乙醇、8-苯胺基-1-萘磺酸、溴化钾、三羟甲基氨基甲烷、甘氨酸 分析纯,北京索莱宝科技有限公司。
ZEN3600 型纳米激光粒度仪 英国马尔文仪器有限公司;LS55 型荧光分光光度计 美国PE 公司;PB-10 型pH 计 赛多利斯(德国)集团;JM-30D 型超声波清洗器 深圳洁盟公司;FDA5-2.5 型冷冻干燥机 北京GOLD SIM 仪器有限公司;UV-1780 型紫外-可见分光光度计 日本岛津实验器材公司;GL-10MD 型大容量高速冷冻离心机 湘仪仪器(湖南)有限公司;Vertex 70 型傅里叶变换红外光谱仪德国Bruker 公司。
1.2 实验方法
1.2.1 原料预处理 将新鲜菇脚在60 ℃鼓风干燥箱中干燥24 h,粉碎后过孔径0.25 mm 的筛,密封后置于干燥器中待用[9]。
1.2.2 金针菇菇脚基本成分测定 水分的测定:参照GB 5009.3-2016《食品中水分的测定》进行;总糖的测定:参照GB/T 15672-2009《食用菌中总糖含量的测定》进行;粗蛋白的测定:参照GB 5009.5-2016《食品中蛋白质的测定》的凯氏定氮法进行;粗纤维的测定:参照GB/T 5009.10-2003《植物类食品中粗纤维的测定》进行;粗灰分的测定:参照GB 5009.4-2016《食品中灰分的测定》进行;粗脂肪的测定:参照GB 5009.6-2016《食品中脂肪的测定》进行。
1.2.3 金针菇菇脚蛋白的提取 碱提酸沉法[16](CK):将金针菇菇脚粉与蒸馏水按照一定料液比混合置于烧杯中,搅拌均匀,室温溶胀30 min 后用1 mol/L NaOH 调节pH 至12.0,40 ℃水浴振荡提取1 h,结束后在4 ℃条件下8000 r/min 离心20 min,用2mol/L HCl 调节上清液的pH 至蛋白等电点(pH3.0),4 ℃沉淀3 h,取出在8000 r/min 条件下离心20 min,将沉淀用蒸馏水反复水洗2~3 次,将沉淀复溶于少量蒸馏水中,调pH 至中性,透析脱盐48 h,真空冷冻干燥得到菇脚蛋白,-20 ℃保存备用。
超声辅助碱提酸沉法[14](US):调节pH 至碱性后,40 ℃、500 W 超声条件下提取20 min,之后继续40 ℃水浴振荡提取40 min,其余步骤同碱提酸沉法。
胶体磨辅助碱提酸沉法[14](CM):调节pH 至碱性后,胶体磨处理10 min,其余步骤同碱提酸沉法。
胶体磨+超声辅助碱提酸沉法[14](CM+US):调节pH 至碱性后,胶体磨处理10 min,40 ℃、500 W超声条件下提取20 min,之后继续40 ℃水浴振荡提取40 min,其余步骤同碱提酸沉法。
冻融辅助碱提酸沉法[15](FT-20 ℃、FT-80 ℃):调节pH 至碱性后,分别置于-20 ℃、-80 ℃冰箱中完全冻结后再解冻,反复冻融三次后,40 ℃水浴振荡提取1 h,其余步骤同碱提酸沉法。
1.2.4 蛋白质提取率测定 采用考马斯亮蓝法测定蛋白提取液中的蛋白含量,根据公式(1)计算蛋白提取率:
1.2.5 持水性和持油性测定 持水性测定:准确称取0.25 g 菇脚蛋白,放入已精确称重并编号的10 mL离心管中,添加5 mL 蒸馏水,涡旋混匀,室温条件下静置1 h,8000 r/min 条件下离心10 min,倒去上清液,倾斜排空上清液测定离心管中剩余物的质量[17]。计算公式如式(2):
式中,M1表示弃去上清液后离心管和蛋白的质量;M2表示初始离心管和蛋白的质量;M 表示蛋白样品的质量。
持油性测定:准确称取0.25 g 菇脚蛋白放入已精确称重的10 mL 离心管中,添加5 mL 玉米油,涡旋混匀,室温条件下静置1 h,8000 r/min 条件下离心10 min,倒去上清液,并倾斜排空,测定离心管中剩余物的质量[17]。计算公式如式(3):
式中,M1表示弃去上清液后离心管和蛋白的质量;M2表示初始离心管和蛋白的质量;M 表示蛋白样品的质量。
1.2.6 起泡性及起泡稳定性测定 准确称取0.25 g菇脚蛋白,添加50 mL 蒸馏水,涡旋混匀后,用0.1 mol/L HCl 或NaOH 溶液调pH 至7.0,用高速分散机在9500 r/min 条件下高速搅打2 min,随后迅速将其倒入量筒中,并且记录上层泡沫体积V0,室温下静置30 min,测定泡沫体积V1[18]。按照公式(4)、(5)计算:
式中,V 表示初始体积量;V0表示搅拌截至时刻的起泡体积;V1表示静置30 min 后的起泡体积。
1.2.7 乳化性及乳化稳定性测定 准确称取0.25 g蛋白样品,添加25 mL 蒸馏水,涡旋振荡,混匀,用0.1 mol/L HCl 和NaOH 溶液调节pH 至7.0,量取15 mL 各溶液转移到离心管中,分别加入5 mL 玉米油,用高速分散机在15000 r/min 条件下高速搅打2 min。
搅拌完成后,立即从管内液面下方2 cm 处吸取50 μL 溶液,加入含有5 mL 0.1% SDS 溶液的离心管中,涡旋振荡后立即测定500 nm 处的吸光值A0。溶液室温放置15 min 后,再次吸取50 μL,按照同样的步骤测定吸光值At[18]。按照公式(6)、(7)计算:
式中,A0表示样品初始吸光值;At表示样品放置15 min 后的吸光值;c 表示蛋白样品浓度;DF 表示稀释倍数;ϕ表示乳化液中油相的比例,本实验中为0.25;t 表示时间。
1.2.8 溶解性测定 用蒸馏水配制2.0 mg/mL 的菇脚蛋白溶液,用0.1 mol/L HCl 或0.1 mol/L NaOH调溶液pH 至7.0。室温条件下磁力搅拌45 min 后,8000 r/min 条件下离心10 min,用考马斯亮蓝法测定上清液中的蛋白含量[19]。按公式(8)计算:
1.2.9 表面疏水性测定 采用ANS 荧光法[20]。配制1.0 mg/mL 的菇脚蛋白溶液,用0.01 mol/L、pH 7.0 的PBS 缓冲液稀释成0.1~1.0 mg/mL 不同浓度的溶液。取各浓度的样品溶液4 mL,分别加入20 µL 8 mmol/L 的ANS 溶液,涡旋混匀,在暗处静置15 min,用荧光分光光度计测定荧光强度。参数为:激发波长390 nm,发射波长为470 nm,狭缝宽度5 nm。以荧光强度为纵坐标和蛋白质浓度为横坐标作图,初始段的斜率即为蛋白质分子的表面疏水性指数。
1.2.10 巯基含量测定 配制20.0 mg/mL 菇脚蛋白溶液,取1.0 mL 蛋白溶液,加入4.0 mL Tris-Gly-10 mol/L Urea 溶液涡旋混合均匀后,再加入50 µL的Ellman 试剂。室温条件下反应15 min 后,测定412 nm 处的吸光值A。同时以不加Ellman 试剂的溶液作为空白对照,测定吸光值。每个样品平行测定三次。测定游离巯基含量时只需用Tris-Gly 缓冲液(0.086 mol/L Tris,0.09 mol/L Gly,4 mmol/L Na2EDTA·H2O,pH8.0)替换测定总巯基时的Tris-Gly-10 mol/L Urea 溶液即可[21]。计算公式如式(9):
式中:73.53 为Ellman 试剂的摩尔消光系数;A412nm表示加Ellman 试剂时样品溶液吸光值;A 表示不加Ellman 试剂时样品溶液吸光值;D 为稀释倍数;C 为蛋白样品的质量浓度(mg/mL)。
1.2.11 傅里叶变换红外光谱测定 准确称取1 mg菇脚蛋白,加入100 mg 溴化钾在研钵中混合碾磨,用压片机进行压片,放于样品架上进行光谱扫描,用溴化钾压片做空白背景,光谱扫描范围设置为400~4000 cm-1,光谱分辨率为4 cm-1,信号扫描累加32 次[22]。使用OMINIC9.0,Peak Fit 软件处理得到的FT-IR 红外光谱图后,对酰胺I 带(1600~1700 cm-1)的数据进行处理。由拟合峰面积得到菇脚蛋白中各个二级结构的相对含量[23]。
1.2.12 Zeta 电位和平均粒径测定 使用纳米激光粒度仪测定菇脚蛋白的Zeta 电位和平均粒径。配制浓度为0.1 mg/mL 的菇脚蛋白溶液,用0.1 mol/L NaOH溶液将蛋白溶液pH 调至7.0,取800 µL 样品溶液加入电位皿,放入样品池中,测量电位,再测量平均粒径,在室温下重复检测3 次。
1.2.13 内源性荧光光谱测定 配制0.1 mg/mL 的菇脚蛋白溶液,利用荧光分光光度计固定 295 nm 激发波长,得到300~450 nm 之间的发射光谱,扫描速度设置为1200 nm/min,狭缝宽度设置为5 nm,得到蛋白质的内源性荧光光谱[24]。
1.3 数据处理
所有实验数据均测定3 个平行,结果以平均值±标准差表示。采用Excel 和SPSS 20.0 软件对数据进行处理和统计分析,采用Origin 2023 进行制图。采用方差分析中的Tukey 检验分析不同组的差异显著性,P<0.05 时差异显著。
2 结果与分析
2.1 金针菇菇脚基本成分
由表1 可知,菇脚中的总糖含量较高,且脂肪含量很低,金针菇菇脚的蛋白质含量可以达到12.45%(干基),金针菇子实体中的蛋白质含量约为15%~25%[25]。

表1 金针菇菇脚基本成分(%)Table 1 Nutritional compositions of flammulina velutipes (%)
2.2 不同提取方式对菇脚蛋白提取率的影响
由图1 可知,胶体磨联合超声辅助提取组具有最高的蛋白提取率,可以达到39.57%,其次是胶体磨组和超声组,都显著高于对照组,但冻融辅助组的蛋白提取率降低。这可能是由于超声和胶体磨的机械作用能够促进蛋白质的溶出,从而提高了提取率[26],许凤等[14]得出了相似的结论。反复冻融可能使蛋白质不易溶出,因此提取率下降。
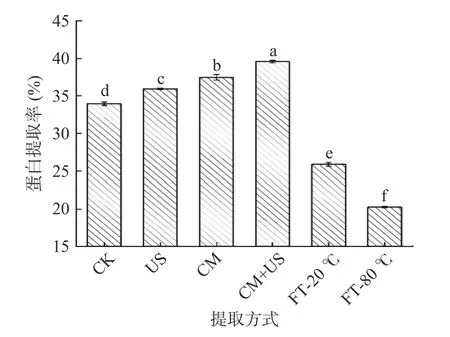
图1 不同提取方式对金针菇菇脚蛋白提取率的影响Fig.1 Effects of different extraction methods on the extraction rate of flammulina velutipes protein
2.3 不同提取方式对菇脚蛋白持水性和持油性的影响
由图2 可知,冻融-80 ℃辅助提取组的持水性和持油性均最高,联合辅助提取组的持水性有所降低,其他处理组之间的持水性和持油性差异不显著,这可能是由于反复冻融冰晶反复形成使蛋白内部空隙变大,具有更好的持水性和持油性。
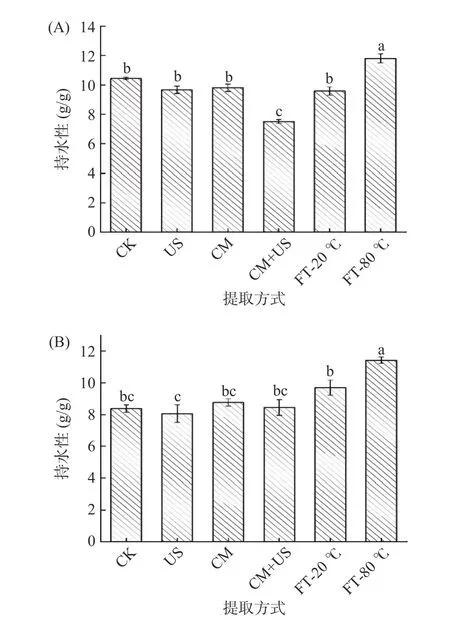
图2 不同提取方式菇脚蛋白的持水性(A)和持油性(B)Fig.2 Oil (A)/water (B) holding capacity of flammulina velutipes stembase protein by different extraction methods
2.4 不同提取方式对菇脚蛋白起泡性、起泡稳定性及乳化性、乳化稳定性的影响
由图3 可知,胶体磨辅助提取以及联合辅助提取组的蛋白质起泡性降低,但具有较高的起泡稳定性,冻融-20 ℃辅助提取组的起泡稳定性降低。这可能是严重的空化效应引起的蛋白质群体变化导致的。空化效应产生的瞬时的高温高压以及胶体磨的高速剪切力可能会破坏蛋白质分子中的基团,导致蛋白质的发泡性能下降。超声组、胶体磨组以及联合辅助提取组的乳化性相比对照组降低,但乳化稳定性提高;冻融处理组的乳化性提高,但乳化稳定性降低。这与Nazari 等[26]的结论相似,高强度超声可能导致蛋白质变性,从而减慢油水界面处的蛋白质吸收,从而降低蛋白质的乳化性。乳化稳定性的增加可能与蛋白质分子更有效地吸附在油水界面上有关,这一变化趋势与溶解性的基本一致。
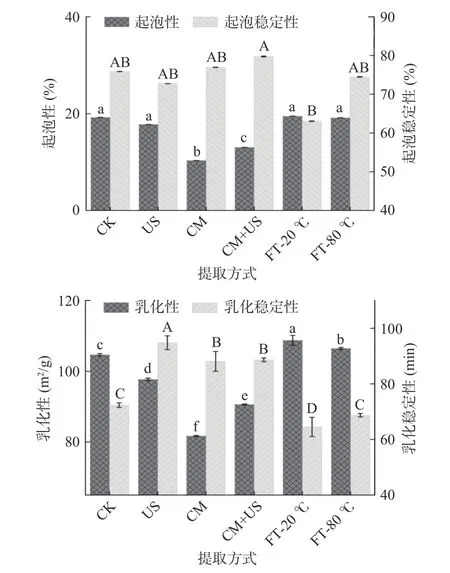
图3 不同提取方式菇脚蛋白的起泡性、起泡稳定性(A)及乳化性、乳化稳定性(B)Fig.3 Foaming capacity and foaming stability (A)/ emulsifying activity (EAI) and emulsifying stability (B) of flammulina velutipes stembase protein by different extraction methods
2.5 不同提取方式对菇脚蛋白溶解性、巯基含量和表面疏水性的影响
从表2 可知,胶体磨辅助提取组和联合辅助处理组溶解性降低,蛋白的溶解性变差,使得在O/W 界面上吸附的蛋白减少,界面薄膜的稳定性被破坏,这也与乳化性和乳化稳定性的降低有关。各处理组的总巯基和游离巯基含量相比对照组都显著(P<0.05)降低。超声辅助提取组、胶体磨辅助提取组以及联合辅助提取组的表面疏水性显著高于对照组,冻融组的表面疏水性显著(P<0.05)降低。表面疏水性是用于评估暴露于蛋白质表面的疏水基团数量的一种方法,通常用于反映蛋白质结构的变化。超声和胶体磨的空化效应和剪切作用会破坏蛋白质的分子结构,导致蛋白质的结构发生重排,其中更多的疏水性基团暴露,增加了蛋白的表面疏水性[27]。游离巯基参与二硫键的形成,对于稳定蛋白质结构有重要作用。超声波的空化效应会使空腔的气相中产生瞬态的自由基,彼此之间会交叉反应产生过氧化氢。这些过氧化氢能与蛋白质分子发生反应,导致蛋白质分子的结构发生改变,一些敏感的官能团也会被氧化,其中就包括游离的巯基。通常,巯基主要存在于蛋白质分子的疏水区中。超声和胶体磨提取组的菇脚蛋白,其分子结构发生改变后,内部的游离巯基就会暴露出来,更多的游离巯基被氧化,所以含量有所减少,这与Fu 等[28]的结论相似。
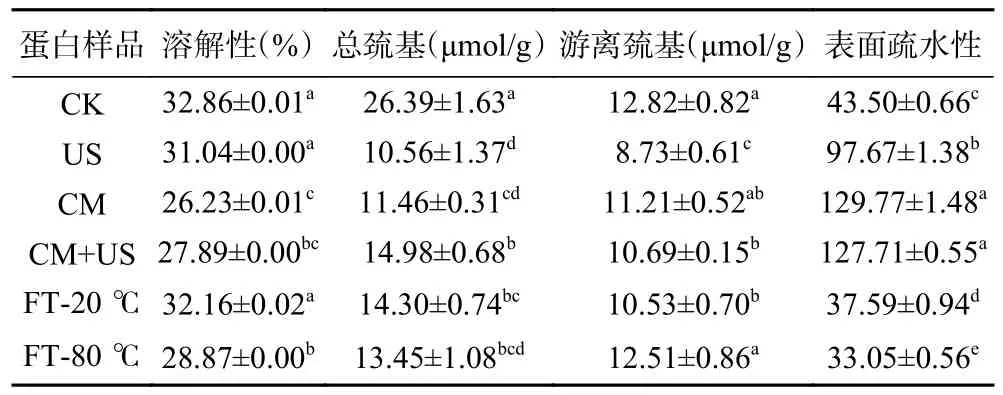
表2 不同提取方式菇脚蛋白的溶解性、巯基含量和表面疏水性Table 2 Solubility,sulfhydryl content and surface hydrophobicity of flammulina velutipes stembase protein
2.6 不同提取方式对菇脚蛋白红外光谱的影响
从图4 可以看出,不同提取方式得到的金针菇菇脚蛋白均具有酰胺A、B 和酰胺Ⅰ、Ⅱ、Ⅲ的特征吸收峰。有几个特征峰吸收强度有所不同,说明不同菇脚蛋白的氨基酸残基存在不同。其中,酰胺A 带主要是由 N-H 键的伸缩振动引起,N-H 伸缩振动发生的范围是 3400~3450 cm-1,但当 N-H 基团参与氢键形成时,其振动频率会向右偏移,各提取组的酰胺A 带均位于3300 cm-1左右,说明有较多的 N-H 基团参与氢键形成,2900 cm-1左右的强吸收峰是由酰胺B 带CH2基团的反对称收缩振动产生的;1650~1700 cm-1的吸收峰是由酰胺Ⅰ带C=O 的伸缩振动产生的,其中,超声辅助组和胶体磨辅助以及联合处理组的吸收峰强度相比对照组显著增强,这是因为C=O 的伸缩振动发生了改变;1500~1550 cm-1的吸收峰是酰胺Ⅱ带的C-N 伸缩振动和COO-伸缩振动产生的;1230~1260 cm-1的吸收峰是由于酰胺Ⅲ带的C-N 伸缩振动和N-H 弯曲振动。
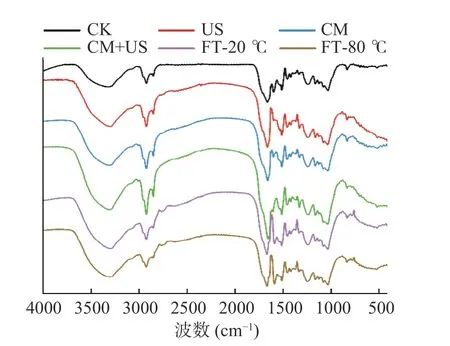
图4 不同提取方式菇脚蛋白傅里叶变换红外光谱图Fig.4 Fourier transform infrared spectra of flammulina velutipes stembase protein
2.7 不同提取方式对菇脚蛋白二级结构的影响
从表3 中可以看出,大多处理组的α-螺旋含量为0,在酸性条件下α-螺旋相对含量低可能是因为蛋白分子间电荷发生中和反应,产生静电作用,影响氢键的稳定性;超声辅助组、胶体磨辅助组以及联合辅助组与对照组相比,β-折叠显著(P<0.05)减少,β-转角显著增加;而冻融辅助组的β-折叠、β-转角含量具有相反的变化趋势。这是因为超声波的空化效应以及胶体磨的高速剪切作用,破坏了氢键和静电相互作用这些非共价键相互作用,导致β-折叠的含量降低,并同时使β-转角的含量升高;而反复冻融破坏了维持蛋白质二级结构的氢键,使得蛋白质的结构发生了变化,部分有序结构变为无序构象,Xu 等[20]研究具有相似的结果,蛋白质二级结构中β-折叠含量的减少表明负责疏水相互作用的蛋白质位点暴露,这也会使蛋白质分子的表面疏水性增加。
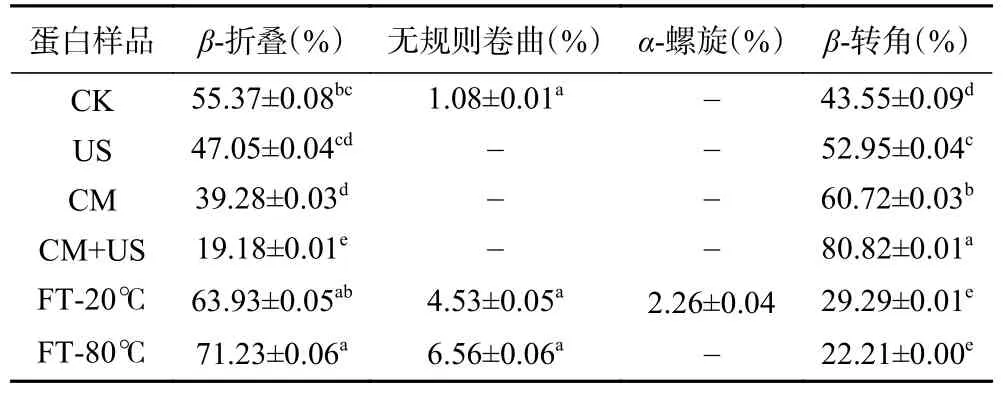
表3 不同提取方式菇脚蛋白二级结构含量Table 3 Secondary structure content of flammulina velutipes stembase protein
2.8 不同提取方式对菇脚蛋白粒径和电位的影响
图5 显示,各物理辅助提取组的粒径相比对照组显著(P<0.05)减小。可能是由于超声的空化作用会产生瞬时的高温高压、胶体磨的高速剪切作用以及反复冻融破坏了蛋白质的非共价键相互作用,比如氢键、静电相互作用和疏水相互作用,会使得一些较大的蛋白质聚集体解离,产生更小的蛋白质分子,从而平均粒径减小,这提高了体系稳定性,与其起泡稳定性和乳化稳定性的提高相互印证。各物理辅助提取组的Zeta 电位绝对值相比对照组显著增加。Zeta 电位绝对值较高的体系,表明分子间的排斥力较强,彼此之间不易形成聚集体,这种体系相对来说比较稳定,反之则体系不稳定[29]。先前隐藏在蛋白结构中的带电离子经过超声和胶体磨以及反复冻融处理后会相互排斥,表面所带的负电荷越多,从而使Zeta 电位绝对值增加,更多的同性电荷间的相互排斥会使蛋白质溶液更稳定,蛋白的聚沉减少[30]。
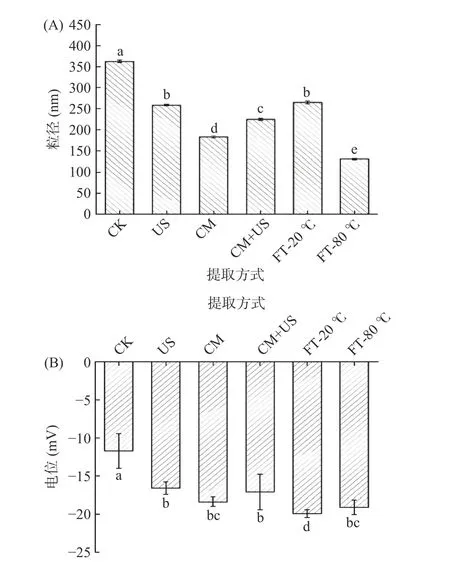
图5 不同提取方式对菇脚蛋白粒径(A)和电位(B)的影响Fig.5 Effects of different extraction methods on the particle size (A) and zeta potential (B) of flammulina velutipes stembase protein
2.9 不同提取方式对菇脚蛋白荧光光谱的影响
蛋白质的荧光光谱反映了蛋白质的构象变化,可以改变色氨酸、酪氨酸和苯丙氨酸基团的局部分子环境。如图6 所示,超声组和胶体磨组的荧光强度明显增强,这可能是由于超声和胶体磨处理的过程中产生的高剪切力,蛋白质分子内部的疏水相互作用被破坏,会暴露出更多蛋白质分子内部的疏水基团,从而引起荧光强度的增强[31]。但联合辅助提取组和冻融组的荧光强度有所下降,这可能是因为过度的剪切力使得一些小的蛋白质颗粒通过疏水相互作用又再次形成大的蛋白质聚集体,一些氨基酸残基暴露出来,相互作用增强,从而产生荧光猝灭,进而使荧光强度降低[32-33]。
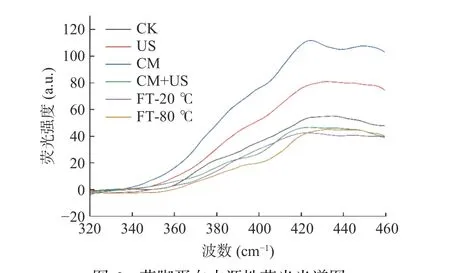
图6 菇脚蛋白内源性荧光光谱图Fig.6 Endogenous fluorescence spectrum of flammulina velutipes stembase protein
3 结论
本研究对比了不同提取方式提取的金针菇菇脚蛋白的理化、功能和结构性质之间的差异。结果表明,超声辅助碱提、胶体磨辅助碱提可以显著(P<0.05)提高菇脚蛋白的提取率,并能在一定程度上改善菇脚蛋白的功能性质,乳化稳定性和起泡稳定性显著(P<0.05)提高;二级结构中β-折叠含量显著(P<0.05)降低,β-转角含量显著(P<0.05)增加,这些结构性质的变化与表面疏水性和稳定性等理化性质的提高密切相关。二者联合辅助提取虽然具有最高的提取率,但由于其过度的剪切力会导致蛋白的乳化性和起泡性等功能性质显著降低;冻融辅助提取具有相对较低的提取率,并且会降低菇脚蛋白的表面疏水性和巯基含量,其最大缺点是需要时间较长,而且需要冷冻设备且能耗较大。基于此,超声和胶体磨辅助提取可以作为一种高效率提取金针菇菇脚蛋白的方法,在金针菇副产物的综合利用方面具有重要意义。
© The Author(s) 2024.This is an Open Access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by-nc-nd/4.0/).
猜你喜欢
意林·全彩Color(2018年9期)2018-10-12
中国计划生育学杂志(2017年3期)2017-06-01
水利科技与经济(2017年2期)2017-04-22
农产品市场周刊(2016年12期)2016-05-24
农家科技中旬版(2016年12期)2016-04-16
食药用菌(2016年6期)2016-03-01
橡胶工业(2015年2期)2015-07-29
河北大学学报(自然科学版)(2015年1期)2015-02-27
生殖医学杂志(2013年4期)2013-03-11
生殖医学杂志(2013年3期)2013-03-11
