
克罗恩病和牙周炎的共有免疫相关基因及其作为诊断生物标志物的潜力
2024-04-16 06:15张志豪王月秋孙思怡
口腔医学 2024年3期
张志豪,杨 益,王月秋,孙思怡,陈 虹,舒 菲,刘 梅
克罗恩病(Crohn’s disease,CD)是一类病因不明的肠道慢性非特异性炎性疾病,目前认为CD可能是基因脆弱人群中免疫细胞对特定菌群异常反应的结果[1],遗传、免疫和环境因素是其影响因素[2]。CD全球发病率高[3],主要表现为慢性腹痛和排便习惯改变,一般还伴有眼睛、口腔、皮肤、关节等部位的病变[4]。肠道微生物群落紊乱,即肠道生态失调,是CD的一个明显标志[5]。
牙周炎和CD具有一定的相关性,CD患者牙周炎的患病率相对更高[6-7],且伴发的口腔唾液功能受损[8]在一定程度上也促进了牙周炎的发生发展[9]。CD患者的口腔菌群和肠道菌群较为相似,有研究发现牙龈卟啉单胞菌(Porphyromonasgingivalis)、具核梭杆菌(Fusobacteriumnucleatum)等牙周致病菌在CD患者的肠道黏膜组织中富集[10-11]。此外,牙周炎和CD在疾病发展过程中,许多共同的细胞因子及免疫反应参与了组织损伤[6,12]。本研究旨在评估牙周炎和CD的共有免疫相关基因及其作为诊断标志物的价值,为探索牙周炎与CD的潜在联系提供研究依据。
1 资料与方法
1.1 数据获取与处理
CD和牙周炎的表达数据来自Gene Expression Omnibus(GEO)数据库(https://www.ncbi.nlm.nih.gov/geo/)。纳入标准为:①采用阵列法生成基因表达谱;②牙周炎数据集样本来自牙龈组织,CD数据集样本来自肠道组织;③数据集包含对照组样本;④样本来源为智人。牙周炎数据集GSE10334、GSE16134均基于GPL570[HG-U133_Plus_2]Affymetrix人类基因组U133 Plus 2.0阵列生成,其中GSE10334包含183个牙周炎牙龈样本和64个健康牙龈样本[13],GSE16134包含241个牙周炎牙龈样本和69个健康牙龈样本[14]。CD数据集GSE126124、GSE75214均基于GPL6244[HuGene-1_0-st]Affymetrix 人类基因组1.0 ST阵列生成,其中GSE126124包含37个CD肠道组织样本和21个健康肠道组织样本[15],GSE75214包含59个CD肠道组织样本和22个健康肠道组织样本[16]。对所有数据集进行标准化,根据临床信息区分疾病组与健康组,并将数据集中的信息与智人基因组进行对比后,匹配数据集中的探针ID与基因名称,将处理完的数据输出为CD和牙周炎处理后的数据集。所有的数据分析均使用R软件(4.2.1)进行。
1.2 差异表达分析
在将原始数据处理之后,使用R语言(4.2.1)的limma包(3.52.4版本)[17]对牙周炎和CD数据进行差异分析(differential expression analysis,DEA)。筛选阈值设置为P<0.05同时|logFC|>0.5,将符合筛选条件的基因定义为差异基因(differentially expressed genes,DEGs)。通过DEA分别筛选出牙周炎和CD中表达异常的DEGs,并使用R语言(4.2.1)的“ggplot2”包(3.3.6版本)以火山图模式分别展示牙周炎和CD中数据集中的DEGs[18]。然后将在牙周炎数据集和CD数据集中同时出现的DEGs定义为共同DEGs(overlapping DEGs),使用R语言(4.2.1)的“ggVennDiagram”包(1.2.2版本)绘制牙周炎和CD共同DEGs的Veen图。
1.3 牙周炎与CD共同DEGs的功能富集分析
为了研究共同DEGs在牙周炎和CD中发挥的主要功能,使用R语言(4.2.1)的“clusterProfiler”包(4.4.4版本)[19]和“DOSE”包(3.24.2版本)[20]对其进行GO富集分析(Gene Ontology enrichment analysis)[21]、KEGG富集分析(Kyoto Encyclopedia of genes and ggenomes pathway enrichment analysis)[22]和DO富集分析(Disease Ontology enrichment analysis),其中GO富集分析的术语包括生物过程(BP)、细胞成分(CC)和分子功能(MF)。将GO分析、KEGG分析、DO分析的阈值均设置为P<0.05。使用“ggplot2”包(3.3.6版本)以气泡图和条形图模式展示富集分析结果。
1.4 构建PPI网络
为了分析共同DEGs之间的相互作用,使用STRING数据库(11.5版本)构建共同DEGs的PPI网络图[23],将“minimum required interaction score”设置为0.4,并使用Cytoscape软件将网络图可视化[24]。在PPI网络中,每个节点都代表这一个相应基因编码的蛋白质,使用Cytoscape的MCODE插件筛计算每个节点的连通度[25],连通度代表了与节点相关的其他节点数量,将节点顶部的基因作为hub gene。节点越大,颜色越深,代表对应基因的联通度越高。
1.5 加权基因共表达网络分析
使用R语言(4.2.1)中的“WGCNA”包(1.71版本)进行加权基因共表达网络分析[26],按标准差排序筛选出WGCNA表达中的前5 000个基因,查看并删除数据集中的离群值,然后分别选取β=16和β=10作为CD和牙周炎的软阈值构建加权基因共表达模型,并将基因分为不同的模块。将每个模块包含基因的最小值设置为30。通过Pick Soft Threshold函数计算出加权参数的最佳取值,使网络在尽可能保留连通性信息的条件下更接近于无尺度网络,并作为后续网络构建的软阈值。之后依次构建邻接矩阵和拓扑重叠矩阵,构建共表达网络,计算各模块基因与临床性状的相关性,将相关性最高的模块定义为关键模块,并筛选出关键模块中的基因以用于后续的分析。
1.6 关键串扰基因的鉴定
从共同DEGs和关键模块基因中筛选出CD和牙周炎中与疾病相关的串扰基因。使用R语言(4.2.1)的“ggVennDiagram”包(1.2.2版本)绘制牙周炎和CD的串扰基因的Veen图。
1.7 免疫浸润分析
为了明确免疫过程在CD和牙周炎疾病进展中的作用,使用R语言(4.2.1)“GSVA”包(1.46.0版本)中的ssGSEA算法对CD和牙周炎样本进行免疫评估[27],评估结果分为免疫细胞和免疫过程两部分,一共包含16个免疫细胞和13个免疫过程,免疫细胞相关基因从以往研究中获得[28]。使用“corrplot”包(0.92版本)分别计算CD和牙周炎中免疫细胞和免疫功能的相关性,并分析疾病组与健康组之间免疫评分的差异。接着分析串扰基因与免疫细胞及免疫过程的相关性,并选择在CD和牙周炎中与免疫相关最多的基因作为核心基因(core genes)进行后续分析。
1.8 构建并验证诊断模型
针对核心基因,通过Logistic逐步回归分析建立诊断模型[29],使用受试者工作特征(receiver operating characteristic curve,ROC)曲线和曲线下面积(area under curve,AUC)评估诊断模型的准确性,并在独立外部数据集(CD:GSE75214,牙周炎:GSE16134)中验证诊断模型的准确性,结果使用“ggplot2”包可视化。
2 结 果
2.1 CD和牙周炎共同差异基因的识别
在CD数据集(GSE126124)中共筛选出697个DEGs,其中包括410个上调基因和287个下调基因(图1A、C)。在牙周炎的数据集(GSE10334)中共筛选出了1 262个DEGs,其中包括622个上调基因和640个下调基因(图1B、D)。在CD和牙周炎的差异基因中筛选出了114个共同上调DEGs和29个共同下调DEGs(图1E、F),并将它们定义为共同DEGs,共143个。
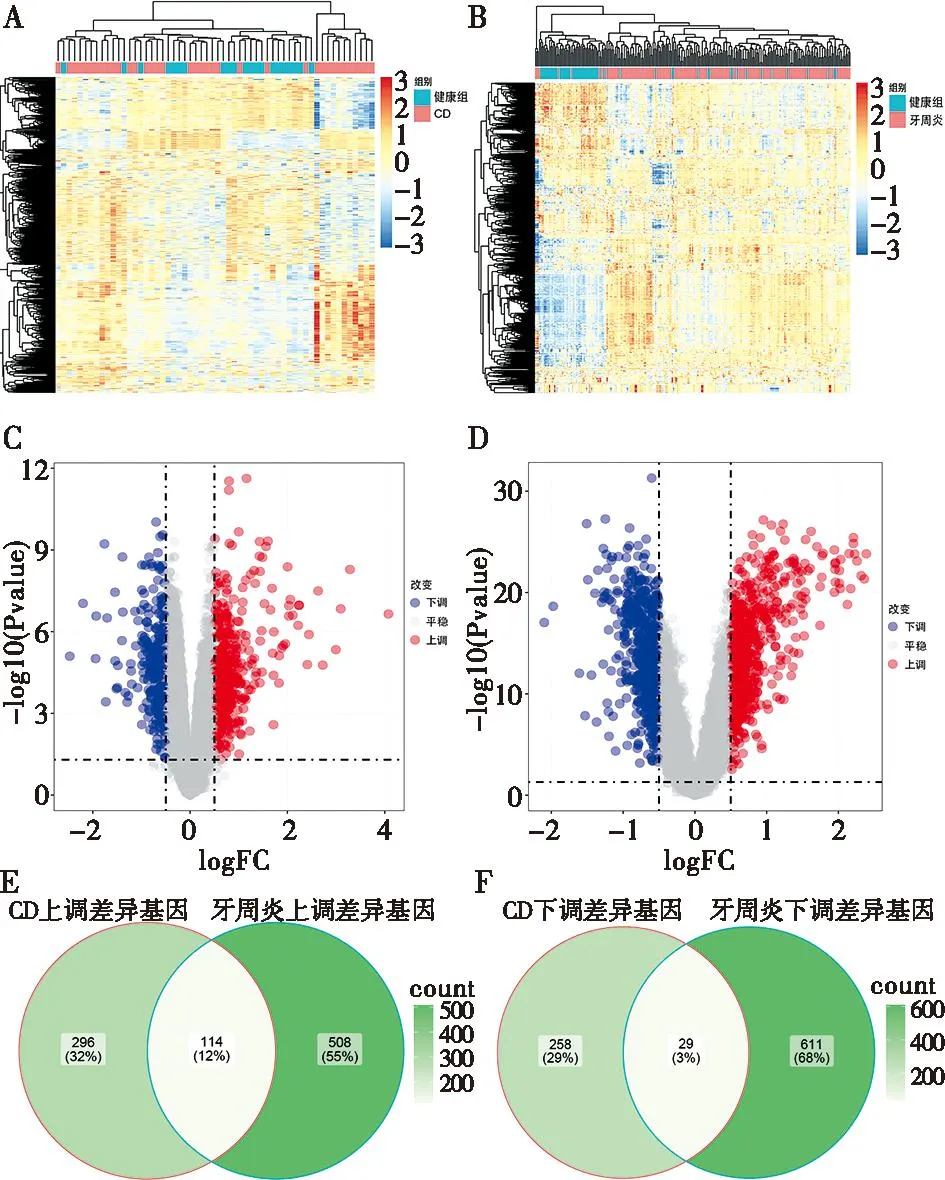
A:GSE126124热图;B:GSE10334热图;C:GSE162124的火山图;D:GSE10334的火山图;E:GSE126124和GSE10334共有上调DEGs;F:GSE126124和GSE10334共有下调DEGs。
2.2 牙周炎与CD共同DEGs的功能富集分析
为了更好地了解共同DEGs的功能,本研究对其进行了功能富集分析,结果如图2所示。GO分析显示共同DEGs主要与白细胞迁移(leukocyte migration)、白细胞趋化反应(leukocyte chemotaxis)、髓白细胞迁移(myeloid leukocyte migration)、免疫反应激活(activation of immune response)、炎症反应调节(regulation of inflammatory response)等生物过程有关(图2A)。

A:GO富集;B:KEGG富集;C:DO分析。
KEGG分析显示,共同DEGs可能与TNF信号通路(TNF signaling pathway)、脂质与动脉粥样硬化(lipid and atherosclerosis)、类风湿性关节炎(rheumatoid arthritis)、IL-17信号通路(IL-17 signaling pathway)有关(图2B)。
DO分析表明共同DEGs主要与动脉硬化(arteriosclerosis)、动脉硬化性心血管疾病(arteriosclerotic cardiovascular disease)、细菌性传染病(bacterial infectious disease)等疾病相关(图2C)。
2.3 构建共同DEGs的PPI网络
使用CD和牙周炎共同DEGs构建PPI网络,其中包括143个节点和664个边缘。图3显示了基于PPI网络拓扑特征分析的前20个节点。根据拓扑特征,白介素(interleukin,IL)-1β、CXC趋化因子配体(CXC chemokine ligand,CXCL)8、IL-6和基质金属蛋白酶(matrix metalloproteinase,MMP)9在生物网络中与其他基因的联通程度最高,可能是影响CD和牙周炎发展的重要基因。
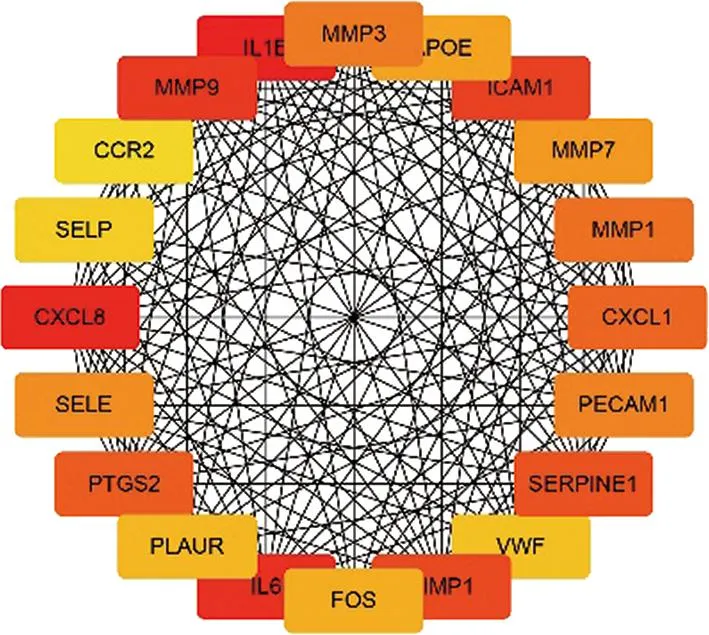
图3 PPI网络(包含重叠度前20基因)
2.4 筛选关键模块
图4A、B分别代表了以CD和牙周炎样本构建的共表达网络,其中CD分为11个模块,牙周炎分为15个模块。Pearson相关系数分析表明,CD与green模块相关性最高(r=0.65,P<0.000 1),该模块包含171个基因;牙周炎与brown模块相关性最高(r=0.61,P<0.000 1),该模块包含332个基因(图4C、D)。使用两模块中的基因进行下一步分析,以筛选关键串扰基因。
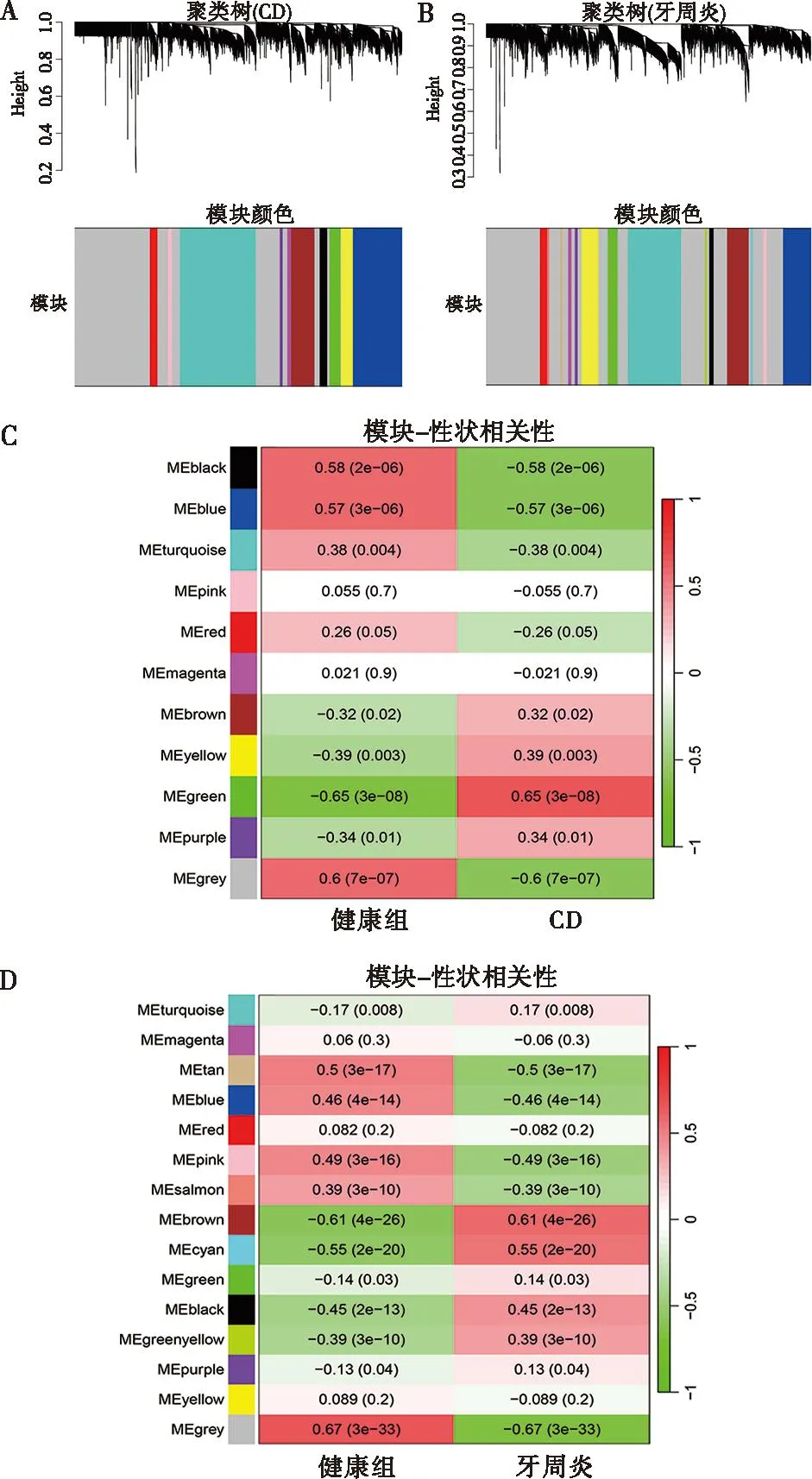
A:CD数据集中标准偏差最高的前5 000个基因的层次聚类树状图;B:牙周炎数据集中标准偏差最高的前5 000个基因的层次聚类树状图;C:CD数据集中的模块-性状相关性;D:牙周炎数据集中的模块-性状相关性。
2.5 关键串扰基因的鉴定
对CD关键模块的基因、牙周炎关键模块的基因和共同DEGs进行分析,图5显示共有11个基因既存在于两种疾病的关键模块中,也存在于二者的共同DEGs中,因此将这11个基因定义为关键串扰基因。
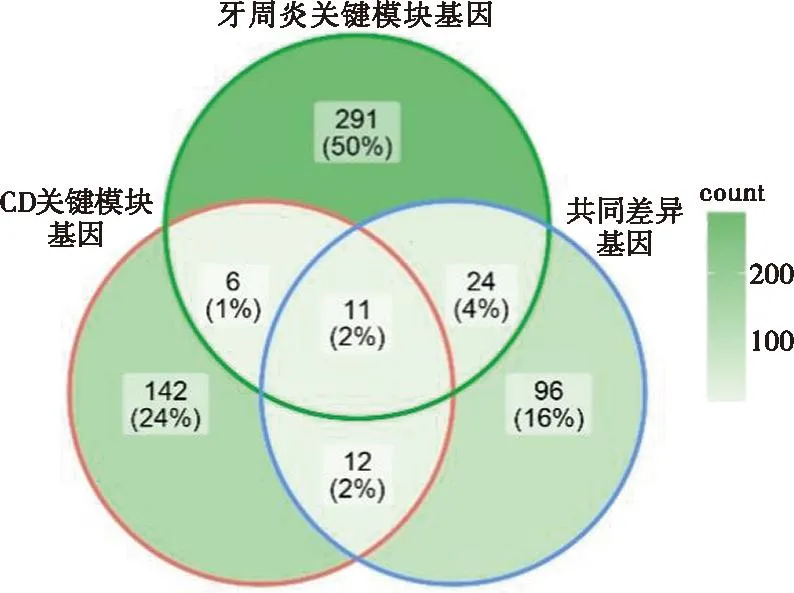
图5 CD和牙周炎关键模块中的重叠DEGs
2.6 免疫浸润分析
富集分析和PPI网络发现免疫反应可能参与了CD和牙周炎的发展,因此使用ssGSEA算法对两个数据集中疾病组与健康组的免疫细胞与免疫过程进行评分,结果表明包括CD8+T细胞、NK细胞、HLA系统参与的免疫过程、CCR家族参与的免疫过程在内的多个免疫细胞与过程的表达存在统计学差异(图6A~D)。此外,本研究还分别分析了11个关键串扰基因在CD数据集和牙周炎数据集中与免疫细胞及免疫过程的相关性(图6E、F),通过对基因与免疫关联程度的分析,发现HLA-DMA、CD38、PIM2、TGM2四个基因从数量以及关联程度两个角度与免疫细胞和免疫过程相关度最高,因此将其定义为核心基因。

A、B:GSE126124数据集中各组免疫细胞和免疫过程的表达;C、D:GSE10334数据集中各组免疫细胞和免疫过程的表达;E:CD数据集中关键串扰基因与免疫细胞及免疫过程之间相关性的热图;F:牙周炎数据集中串扰基因与免疫细胞及免疫过程之间相关性的热图;*:P<0.05;**:P<0.01;***:P<0.001;ns:P>0.05。
2.7 构建并验证诊断模型
以筛选出的核心基因为基础,使用Logistic逐步回归分析建立诊断模型,发现诊断模型在CD和牙周炎数据集中均具有良好的诊断效力;在CD数据集(GSE126124)中AUC=0.873,在牙周炎数据集(GSE10334)中AUC=0.840。为了进一步证明核心基因的诊断效力,在另外两个独立的外部数据集(CD:GSE75214,牙周炎:GSE16134)中进行验证,结果表明在外部数据集中其仍然具有良好的诊断效果,在GSE75214中AUC=0.836,在GSE16134中AUC=0.865。以上结果表明,该模型对CD和牙周炎的诊断具有一定的指导意义。见图7。
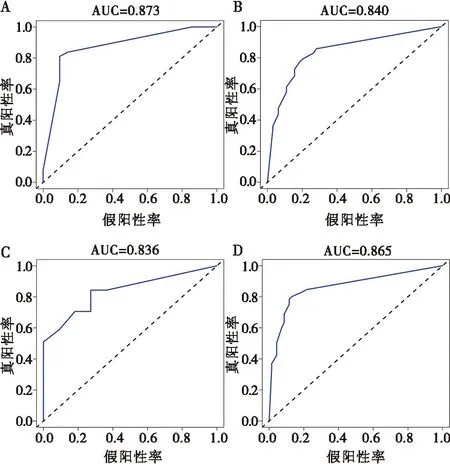
A:GSE126124中诊断模型的ROC曲线;B:GSE10334中诊断模型的ROC曲线;C:GSE75214中诊断模型的ROC曲线;D:GSE16134中诊断模型的ROC曲线。
3 讨 论
CD和牙周炎关系密切且互为影响因素。一项病例对照研究表明,CD患者患有严重牙周炎的概率显著增加[30]。牙周炎患者唾液中的微生物菌群也会加重肠道炎症[31-32]。作为免疫介导的炎症性疾病,CD和牙周炎的致病机制可能存在一些潜在联系,免疫细胞及免疫过程在两种疾病的发生发展中均起到重要作用[6]。因此识别出与免疫相关的串扰基因对更加深入地了解这两种疾病之间的联系具有重要意义,筛选出合适的诊断标志物对疾病的预防、治疗和改善预后也有一定的帮助。
本研究鉴定出143个CD和牙周炎两种疾病的共同DEGs,其中有114个上调基因和29个下调基因,这些基因在TNF信号通路(TNF signaling pathway)、IL-17信号通路(IL-17 signaling pathway)中富集。TNF由免疫系统中的巨噬细胞和T细胞等细胞产生,通过与靶细胞表面的TNF受体结合,激活下游信号通路,参与免疫细胞的活化以响应感染或组织损伤[33]。IL-17信号通路参与防御细菌、真菌等细胞外病原体的免疫反应。IL-17由Th17细胞的T细胞亚群产生,并通过IL-17受体作用于靶细胞[34]。这两条信号通路最终都会导致促炎细胞因子和趋化因子的产生,从而参与免疫过程。TNF通路过度激活与类风湿性关节炎和克罗恩病等炎症性疾病有关[35],IL-17通路失调影响银屑病和多发性硬化症等疾病的发生发展[36]。DO分析表明CD和牙周炎与血管性疾病、细菌感染性疾病有一定程度的相似,GO分析表明免疫在两种疾病中起到重要作用。因此本研究进一步利用WGCNA筛选出与疾病最为相关的基因,与共同DEGs分析后得到了11个串扰基因。
免疫浸润分析发现CD和牙周炎中疾病组和健康组的免疫水平存在明显差别,进一步分析11个串扰基因与免疫的相关性,通过分析各个基因与免疫细胞和免疫过程的相关性,筛选出既在两种疾病中同时与免疫相关程度最高,又与最多数量的免疫细胞和免疫过程相关的4个基因(HLA-DMA、CD38、PIM2、TGM2)。HLA-DMA是HLA复合物的一部分,HLA复合物编码免疫系统识别外来抗原的蛋白质,并参与抗原呈递和T细胞识别的过程,这对激活适应性免疫反应至关重要[37],有研究发现某些HLA等位基因(包括HLA-DMA)与CD和牙周炎易感性之间均存在相关性[38-39]。CD38是一种编码T细胞、B细胞和自然杀伤细胞等免疫细胞表面受体的基因,参与免疫细胞的活化、增殖及细胞因子、趋化因子的产生,在感染或炎症中经常表达上调[40]。Mahanonda等[41]研究发现,CD38在重度牙周炎患者的牙周组织和CD患者肠黏膜组织中均表达增加。CD38可能通过活化T细胞和调节细胞因子的产生在CD和牙周炎的疾病发展中发挥作用。PIM2是一种参与调节细胞生长、增殖和分化的基因[42]。PIM2可通过调节破骨细胞的活性来影响牙周炎的发展。da Silva等[43]发现,PIM2的表达在CD患者的炎症肠道组织中显著上调。TGM2编码的谷氨酰胺转氨酶可以调节免疫细胞功能,TGM2可能通过影响细胞外基质形成和免疫细胞活化,在牙周炎和CD的发病机制中发挥作用[44-45]。
HLA-DMA、CD38、PIM2、TGM2参与抗原呈递及免疫细胞的活化、增殖和分化,任何一种基因表达失调都可能导致免疫功能障碍,继而影响CD和牙周炎的发展。Logistic回归分析是疾病自动诊断中常用的一种广义线性回归分析方法,可以用来预测疾病发生的概率,本研究Logistic回归分析表明,以这4个基因作为基础构建的模型具有良好效力,在一定程度上有助于CD和牙周炎的诊断,同时这些基因还可能作为潜在的治疗靶点。
本研究从免疫角度筛选出了CD和牙周炎的核心基因,为CD和牙周炎的共病机制以及诊断、治疗提供了新思路。但本研究也存在一定的局限性,CD和牙周炎患者是不同的群体,其患者情况不同,可能存在个体差异;且本研究结果是使用生物信息学分析得出的,后续还有待进一步研究验证。
4 结 论
HLA-DMA、CD38、PIM2、TGM2基因参与了CD和牙周炎的发生发展,在免疫反应中扮演着重要角色,以这4个基因构建的诊断模型对两种疾病都有良好的诊断效力。
猜你喜欢
大自然探索(2024年1期)2024-03-19
当代水产(2021年10期)2022-01-12
保健医苑(2021年7期)2021-08-13
昆明医科大学学报(2021年6期)2021-07-31
今日农业(2020年22期)2020-12-14
甘肃科技(2020年20期)2020-04-13
实用口腔医学杂志(2017年6期)2017-09-19
中国继续医学教育(2015年4期)2016-01-07
中国医疗美容(2015年1期)2015-07-12
科技经济市场(2014年5期)2014-09-09
