
基于重测序的23份香菇种质资源全基因组序列分析
2024-04-02 12:15:56宋琳琳陈红芝毋柳柳李畅孟丽孔维丽
中国瓜菜 2024年3期
宋琳琳 陈红芝 毋柳柳 李畅 孟丽 孔维丽

摘 要:為了对河南香菇主产区的主栽品种进行鉴定,并分析其遗传多样性,将河南主栽的23份香菇种质资源进行重测序,对单核苷酸多态性(single nucleotide polymorphism,SNP)和小片段插入缺失(insertion-deletion,InDel)等数据进行统计和分析,并基于SNP变异对23份香菇资源进行遗传结构分析、进化树构建和主成分分析。结果表明,在23份香菇样本中,比对到参考基因组上的reads数目占总数的比例范围为72.53%~90.60%,样品平均测序深度范围为12.83~19.99,覆盖率范围为91.89%~99.32%。SNP位点共计14 115 075个,InDel共计1 909 516个。23份香菇样本的群体遗传结构及系统发育分析表明其包含3个谱系,遗传距离0.054 90~0.656 89,推测它们至少有3个祖先遗传成分。结合主成分分析法明确各菌种间的亲缘关系远近,证明了菌种分支具有明显的地域性。综合分析表明,以基因序列相似度、遗传距离差异及亲缘关系远近为主要依据特点,有助于对香菇地方品种命名和其特征特性关系的认识,促进优异种质资源的交流与利用。
关键词:香菇;重测序;单核苷酸多态性;插入缺失变异;群体遗传结构
中图分类号:S646 文献标志码:A 文章编号:1673-2871(2024)03-028-07
Whole-genome squence analysis of 23 Lentinula edodes germplasms based on genome re-sequencing
SONG Linlin1, CHEN Hongzhi2, WU Liuliu1, LI Chang3, MENG Li1, KONG Weili4
(1. School of Life Science , Henan Institute of Science and Technology, Xinxiang 453003, Henan, China; 2. Xinxiang Institute of Engineering, Xinxiang, 453700, Henan, China; 3. School of Faculty of Arts and Law, Zhengzhou Technology and Business University, Zhengzhou, 451400, Henan, China; 4. Edible Fungus Research Institute, Henan Academy of Agricultural Sciences, Zhengzhou 450000, Henan, China)
Abstract: To identify and analyze the genetic diversity of Lentinula edodes isolated from the main producing area in Henan province, the study resequenced the genomes of 23 cultivated L. edodes germplasm resources. The researchers analyzed the single nucleotide polymorphism (SNP) and insertion-deletion (InDel) data, and finished the genetic structure analysis, phylogenetic tree construction and principal component analysis based on the SNP variation. The proportion of reads matched to the reference genome ranged from 72.53% to 90.60% among the samples, with an average sequencing depth ranging from 12.83 to 19.99 and coverage rates ranging from 91.89% to 99.32%. The researchers identified a total of 14 115 075 SNP sites and 1 909 516 InDel sites. The results revealed the presence of three lineages with genetic distances ranging from 0.054 90 to 0.656 89, indicating the existence of at least three ancestral genetic components. Based on the principal component analysis, the relative distance of each strain was confirmed, which proved that the strain branches had obvious regional characteristics.The comprehensive analysis considered the similarity of gene sequences, genetic distance differences, and the relationships among relatives as the main characteristics. The understanding of these characteristics could facilitate the exchange and utilization of excellent germplasm resources. Additionally, the study provided insights into the relationship between the naming of local varieties and their genetic characteristics.
Key words: Lentinula edodes; Resequencing; Single nucleotide polymorphism; Insertion-deletion variation; Population genetic structure
香菇(Lentinula edodes)又名香蕈、冬菇,隶属于伞菌目(Agaricales)光茸菌科(Omphalotaceae)小香菇属(Lentinula)食用菌,是国际公认的健康食品[1-2]。香菇富含蛋白质、多糖、维生素、微量元素等营养物质。香菇多糖已被证实具有抗病毒、抗衰老、预防心脑血管疾病、预防肝硬化、保护肾功能等功效[3-4];香菇粗纤维可以帮助消化、缓解便秘、减肥;香菇中的嘌呤、胆碱等可以降血压、降血脂,预防肝硬化及动脉硬化的发生;近年来研究证明,食用香菇对减少糖尿病及其并发症,如坐骨神经痛、视网膜炎等均有一定的治疗作用[5-7]。
据中国食用菌学会2022年统计,2021年河南省食用菌总产量576.13万t,位居全国第一。全省食用菌各类品种折合超过57.57亿袋,产值410.37亿元,较2020年增长2.16%,其中香菇以387.04万t的产量位居各种主要食用菌产量之首,占全部食用菌产量的68.07%。品质好、性状稳定的菌种是香菇产业发展的基础,但多年来河南省品种繁多、引种不清、命名混乱、质量标准不统一等问题已经严重影响和制约产业的健康发展。利用形态和生理生化特征对香菇品种进行鉴定,操作繁琐,且结果不一定可靠[8]。基于各种分子标记开展的香菇种质资源亲缘关系鉴定和遗传多样性分析已陆续进行。汪昊等[9]用简单序列重复区间扩增多态性(inter-simple sequence repeat,ISSR)、相关序列扩增多态性(sequence-related amplified polymorphism,SRAP)和目标区域扩增多态性(target region amplified polymorphism,TRAP)标记对我国常见的19个香菇品种进行序列扩增和多样性分析,表明TRAP标记贡献率最高。吴小燕等[10]、肖东来等[11]、宋莹等[12]、郭金英等[13]利用ISSR分子标记对我国常见香菇栽培品种或野生菌株进行聚类分析,结果表明,野生和栽培菌株可明显分开,且名称相似或者同一地区的栽培品种聚为一类。董慧等[14]利用简单重复序列扩增多态性(simple sequence repeat,SSR)标记对我国香菇51个主栽品种进行聚类分析,认为现有的商业品种大多遗传背景相似(遗传相似系数均高于0.60)。Xiang等[15]利用68个InDel和2个SSR标记,对我国2个地区4个香菇居群的遗传变异进行分析,提出地理位置的不同分布是形成我国香菇当前遗传结构的重要因素。沈秀芬[16]用重测序获得的小片段插入缺失(insertion-deletion, InDel)标记对44份野生和地方主栽香菇菌株进行遗传多样性分析,提出了可挑选优质野生菌株和现有主栽菌株进行杂交以获得优良香菇品种的建议。有关香菇全基因组遗传关系分析的研究较多,但是利用重测序获得的单核苷酸多态性(single nucleotide polymorphism, SNP)标记对香菇菌种,尤其是国内主栽品种进行亲缘关系鉴定和遗传多样性分析的研究较少。
笔者针对河南香菇主产区的23份香菇种质资源进行基因组重测序(whole genome resequencing,WGR),一方面利用获得的SNP变异对23份样本进行遗传结构分析、进化树构建和主成分分析,对其亲缘关系进行鉴定,旨在解决主产区香菇菌种命名混乱的问题。另一方面,通过重测序获得香菇样本的SNP变异、InDel变异等数据,利用生物信息学的方法对其特点进行分析,为后期开发香菇重要性状分子标记、研究香菇遗传多样性和分子育种奠定理论基础。
1 材料与方法
1.1 材料
选取河南省南阳、驻马店、三门峡、漯河4个香菇主产区的23份香菇菌株作为供试材料(表1)。2023年4-6月收集河南省食用菌种质资源库、新乡市农业科学院、驻马店市农业科学院、河南科技学院微生物教研室保存的上述河南主产区的香菇品种。
1.2 菌种的活化
2023年7月,在河南科技学院生命科学学院将收集到的23份香菇菌株接种于新的PDA斜面上,28 ℃ 培养活化。选取无污染、长势良好的活化菌种接种于PDA平板上,在培养基和接种菌块之间用灭菌的玻璃纸隔开,便于后期收集菌丝。28 ℃培养一周后,用灭菌的钥勺将玻璃纸上的菌丝轻轻刮下,放入2 mL冷冻管中,立即进行液氮速冻,放入-80 ℃冰箱保存备用。
1.3 DNA提取和文库构建
将收集到的23份香菇菌丝用干冰送样,由武汉菲沙基因信息有限公司进行DNA提取。检验合格的DNA样品通过Covaris破碎机随机打断成长度为350 bp的片段,DNA片段经末端修复、加ployA尾、加测序接头、纯化、PCR扩增等步骤完成整个文库制备。文库质检合格后,把不同文库按照有效浓度及目标下机数据量的需求进行PE150测序。
1.4 测序质检、同参考基因组进行比对
将测序得到的原始图像经识别后,得到的原始数据(Raw reads)需要进一步去除dA、比例高于10%的含N数据、低质量数据等,得到过滤后的数据(Clean reads)。通过比对Clean reads和Raw reads,以及Q20、Q30的數值等检测测序质量。以香菇Lentinula edodes ASM1547640v1(NCBI Accession:GCA_015476405.1)为参考基因组,将过滤后的Clean reads用微生物重测序分析软件BWA(Burrows wheeler alignment,0.7.17)与参考基因组比对,利用GATK(Genome analysis toolkit,4.2.0.0)软件对PCR重复进行去重处理。对去重后的数据进行比对率、覆盖度和测序深度的统计。
1.5 基因组变异检测
利用GATK软件对样本数据进行检测和过滤,获得样本的SNP和InDel变异信息,并利用ANNOVA软件对香菇全基因组SNP进行注释,获得变异位点发生的位置及类型。
1.6 群体分析
在上述获得的SNP信息基础上,对23份香菇样本进行群体分析。为保证后续群体分析的可靠性,利用Vcftools(0.1.17)软件首先对样本数据进行群体过滤。过滤后群体SNP数量由1 320 359个变为674 565个。利用treebest(1.9.2)构建系统进化树(phylogenetic tree),利用plink(1.90)进行主成分分析(principal components analysis),利用admixture(1.3.0)进行群体遗传结构分析(population structure analysis)。
2 结果与分析
2.1 23份香菇样本的基因重测序分析
利用Illunima平台对23份香菇菌株菌丝DNA进行重测序。Raw reads范围是6 115 825~9 056 198,经过滤后的Clean reads范围是5 436 754~7 857 618,比率为86.77%~92.58%;Rawbases范围是0.92~1.36 G,经过滤后的Clean bases范围是0.80~1.15 G。Q20范围为98.13%~98.82%,Q30范围为91.16%~100%,说明文库构建质量符合样本后续重测序要求。对23份香菇菌株重测序数据与参考基因组进行比较,比对到参考基因组上的reads数目占总数的比例范围为72.53%~90.60%,样品平均测序深度范围是12.83~19.99,覆盖率范围是91.89%~99.32%,说明测序数据基本覆盖参考基因组,可进行后续分析。
2.2 23份香菇菌种的重测序SNP变异和InDel变异分析
23份样本的SNP位点变异范围是307 684~590 751,其中转换变异范围是224 670~431 249,颠换变异范围是80 536~157 657,Ts/Tv平均值为2.734。InDel变异范围是40 421~83 544,其中插入变异范围是21 207~44 368,缺失变异范围是32 842~63 681。在23份香菇种质资源的SNP变异类型中,编码区变异占比最高(30.3%),而编码区基因的变异可能会导致氨基酸序列的改变,从而会引起性状的改变。本试验所统计的外显子变异中,同义突变占59.9%,非同义突变占39.4%。23个菌株中,LE14(香泌阳18-2)外显子变异最大,其次是LE45(南山1号)、LE31(武香1号)和LE38(香18),而这几个品种分别来自泌阳和舞阳。LE12(香9608)外显子变异最小,其次是LE44(9608)、LE10(香LS-1)、LE35(808)、LE41(申香215)和LE4(香808),而这几个品种分别来自舞阳、三门峡和泌阳。
2.3 群体分析
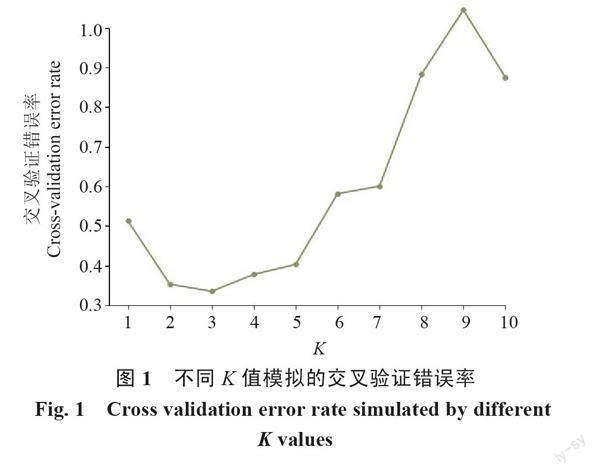
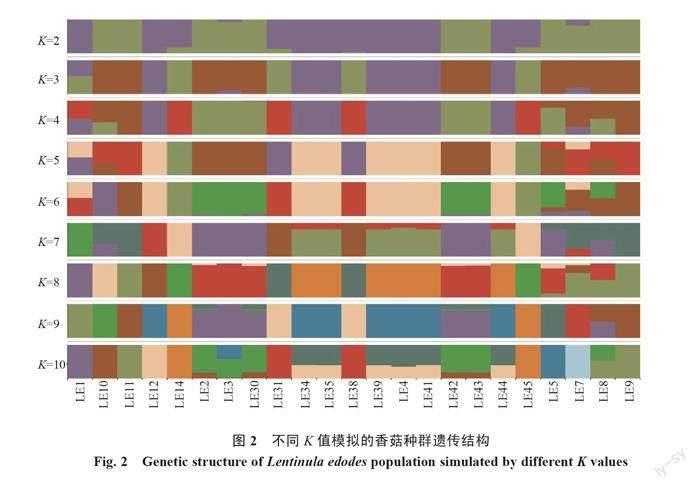
2.3.1 群体遗传结构分析 基于SNP的数据信息,对23份香菇菌株样本进行群体结构分析。由图1可以看出,当K=3时,错误率最低,因此K=3时模型较为可靠。由图2可知,当K=2时,23份香菇菌株样本分化为2个谱系种群;当K=3时,2个谱系种群进一步分化为3个谱系:由棕色构成的第一谱系,分别是9608(LE9)、庆元2号(LE11)、L18(LE8)、香LS-1(LE10)、L26(LE30)、武香1号(LE43)、庆科212(LE42)、灵仙1号(LE2)、L18(LE5),这些香菇大多数来自于西峡、卢氏和泌阳;由紫色构成的第二谱系,分别是香808(LE4)、香9608(LE12)、808(LE35)、武香1号(LE44)、808(LE39)、L9608(LE34)和申香215(LE41),这些香菇来自于河南菌种资源库、泌阳和舞阳;由绿色构成的第三谱系,分别是香泌阳18-2(LE14)、武香1号(LE31)、香18(LE38)、南山1号(LE45),这4株香菇来自泌阳和舞阳。LE1号香菇来源于河南省菌种资源库,推测其由2个祖先遗传成分;LE3和LE7含有2个相同的祖先遗传成分。总的来说,23份香菇菌株有3个谱系,推测至少有3个祖先遗传成分。
2.3.2 主成分分析 基于SNP结果,采用主成分分析方法对23份香菇菌株资源3个群体(pop1、pop2、pop3)的遗传背景相似性进行归类(图3)。通过主成分分析,共提取到2个主成分(PC1:47.26%、PC2:32.37%),方差累积贡献率为79.63%,其能够较好解释变量的总体变化情况。由图3可知,结合PC1和PC2把23份香菇菌种分为三大类,第一类(pop1)为9608(LE9)、庆元2号(LE11)、L18(LE8)、香LS-1(LE10)、L26(LE30)、武香1号(LE43)、庆科212(LE42)、灵仙1号(LE2)、L18(LE5)、香升龙1-1(LE3)、香931-2(LE7),主要由来自西峡、卢氏和泌阳的菌株构成;第二类(pop2)为香931(LE1)、武香1号(LE31)、香18(LE38)、香泌阳18-2(LE14)、南山1号(LE45),主要由来自泌阳和舞阳的菌株构成;第三类(Pop3)为香808(LE4)、香9608(LE12)、808(LE35)、武香1号(LE44)、808(LE39)、L9608(LE34)和申香215(LE41),由來自泌阳和舞阳的菌株构成。供试的23份香菇菌种分类与聚类分类结果基本一致,结果表明,PC1和PC2基本能明显区分这23个品种。主成分分析结果进一步验证了这些菌株亲缘关系非常近,尤其是来自西峡的9608(LE9)和来自卢氏的庆元2号(LE11),来自西峡的灵仙1号(LE2)和L18(LE5),来自泌阳的武香1号(LE43)、庆科212(LE42)和来自舞阳的L26(LE30),来自泌阳的香泌阳18-2(LE14)和来自舞阳的南山1号(LE45),遗传距离几乎重叠。
2.3.3 聚类分析 根据treebest软件计算供试菌株序列间的遗传距离,23份香菇菌株的遗传距离在0.054 90~0.656 89之间。其中,武香1号(LE31)与庆元2号(LE11)的遗传距离最远;L26(LE31)、香18(LE38)、香931(LE1)、香泌阳18-2(LE14)、南山1号(LE45)与其他菌株的遗传距离在0.520 14~0.656 89之间,遗传距离相对较远。
为探究各香菇菌株间的进化关系,将香菇序列构建 NJ 系统进化树(图4),根据物种亲缘关系,23份香菇菌种被细分为3个亚科。南山1号(LE45)、香泌阳18-2(LE14)、香931(LE1)、香18(LE38)、武香1号(LE31)聚为一支,其中,南山1号(LE45)、香泌阳18-2(LE14)2个菌株组成姐妹群,且与香931(LE1)亲缘关系较近;香9608(LE12)、香808(LE4)、808(LE35)、9608(LE44)、808(LE39)、申香215(LE41)、L9608(LE34)7个菌株组成一个单系类群,亲缘关系较近;其余11个品种为第3支,其中,L26(LE30)、庆科212(LE42)2 个菌株组成姐妹群,亲缘关系较近。
3 讨论和结论
在食用菌研究中,传统的分子标记如SSR、ISSR、SRAP和TRAP等常被用于菌株鉴定和种质资源多样性分析。随着高通量测序技术的发展,越来越多的物种基因组被测序和注释,基于全基因组重测序技术开发的SNP[17]、InDel等分子标记也得到广泛应用。笔者的研究针对河南香菇主产区的23份香菇样本进行重测序,通过生物信息学分析,获得了大量的群体变异信息,这为研究香菇遗传多样性提供了理论基础;基于这23份样本的SNP位点变异,笔者进行了遗传结构分析、聚类分析和主成分分析,旨在探索菌株之间的亲缘关系和进行菌种鉴定,为解决河南主产区香菇菌种命名混乱的问题和明确今后分子育种的方向提供了一定的依据。群体遗传结构分析结果表明:23份香菇菌株来源于3个谱系,遗传背景相对单一,这与董慧等[14]和Li等[18]提出的中国现有的香菇商业品种大多遗传背景相似的观点相一致,在今后的育种工作中应尽量拓宽遗传基础,如收集野生香菇菌种资源与现有优良栽培品种进行杂交[16]。聚类分析结果表明,来自于西峡和卢氏的品种多数聚为一类,来自于泌阳和舞阳的品种多数聚为一类,来自泌阳、舞阳的9608和808多数聚为一类,这与吴小燕等[10]提出的名称相似或者同一地区的栽培品种会聚为一类的观点相符合;相近地区的菌种之间遗传分化和遗传距离相对较小,与董慧等[14]和Zhang等[19]的研究结果一致。崔筱等[20]通过香菇菌丝拮抗试验证实来源于西峡、泌阳、卢氏等基地的香菇命名虽然不同(包括香9608、庆元2号、香931-2、香939、香升龙1-1、灵仙1号等),但这些菌株的菌丝无拮抗现象,在笔者的试验中,主成分分析结果进一步验证了这些菌株亲缘关系非常近,尤其是来自西峡的9608(LE9)和来自卢氏的庆元2号(LE11)、西峡的灵仙1号(LE2)和L18(LE5)、泌阳的武香1号(LE43)和庆科212(LE42)、泌阳的香泌阳18-2(LE14)和来自舞阳的南山1号(LE45)。因此初步判断这些香菇品种存在同种异名现象;但来自舞阳的武香1号和来自泌阳的武香1号遗传距离较远,初步判断为同名异种。
防止菌种混乱这一问题,需要从管理者、生产者、推广者等多方进行监督,一是管理部门需通过各种宣传手段增强育种工作人员知识产权保护意识,通过法律法规切实保护食用菌新品种新菌株;二是菌种生产者需自觉树立良好职业道德和严格的执业态度,不得随意命名和编号;三是要提升菌种生产人员的专业技能,严格从业人员考核制度;四是推广者需做好引种溯源工作,切实落实各项食用菌菌种管理制度。
综上所述,基于重测序的SNP变异数据,将河南省香菇主产区的23份资源分为3个谱系,推测它们至少有3个祖先遗传成分,结合主成分分析法明确了各菌种间的亲缘关系远近,证明了菌种分支具有明显的地域性。研究结果有助于对香菇地方品种命名和其特征特性关系的认识,对优异种质资源的交流与利用具有重要的促进作用。
参考文献
[1] CHIKARI F,HAN J,WANG Y,et al.Synergized subcritical-ultrasound-assisted aqueous two-phase extraction,purification,and characterization of mushroompolysaccharides[J].Process Biochemistry,2020,95:297-306.
[2] HU D H,CHEN W,LI X S,et al.Ultraviolet irradiation increased the concentration of vitamin D2 and decreased the concentration of ergosterol in shiitake mushroom(Lentinus edodes)and oyster mushroom(Pleurotus ostreatus)powder in ethanol suspension[J].Acs Omega,2020,5(13):7361-7368.
[3] 曹賢,邹明,高俊峰,等.基于氨基酸含量分析14类香菇的品质特性[J].中国瓜菜,2023,36(8):48-55.
[4] 孙佳星.香菇栽培菌株遗传多样性及对温度的响应与适应性[D].辽宁大连:辽宁师范大学,2022.
[5] JEFF I B,LI S S,PENG X X,et al.Purification,structural elucidation and antitumor activity of a novel mannogalactoglucan from the fruiting bodies of Lentinus edodes[J].Fitoterapia,2013,84:338-346.
[6] LIN Y Y,ZENG H Y,WANG K,et al.Microwave-assisted aqueous two-phase extraction of diverse polysaccharides from Lentinus edodes:Process optimization,structure characterization and antioxidant activity[J].International Journal of Biological Macromolecules,2019,136:305-315.
[7] LEE S,BAE H,KIM N,et al.Optimization of growth conditions of Lentinus edodes mycelium on corn processing waste using response surface analysis[J].Journal of Bioscience and Bioengineering,2008,105(2):161-163.
[8] 卓英,谭琦,陈明杰,等.香菇主要栽培菌株遗传多样性的AFLP分析[J].菌物学报,2006,25(2):203-210.
[9] 汪昊,牛玉蓉,荣成博,等.19个香菇菌株基于ISSR?SRAP和TRAP分子标记的遗传多样性分析[J].江苏农业科学,2019,47(17):54-59.
[10] 吴小燕,鲍红春,李小雷,等.26个香菇菌株的遗传多样性ISSR分析[J].种子,2023,42(5):63-67.
[11] 肖东来,张迪,林衍铨,等.20株香菇菌株的ISSR和F-MSAP分析[J].福建农业学报,2018,33(8):794-798.
[12] 宋莹,刘俊杰,刘岩岩,等.辽宁省主栽香菇菌株ISSR遗传差异性分析[J].北方园艺,2017(5):82-85.
[13] 郭金英,宋彦龙,李超,等.四十个野生香菇菌株遗传多样性分析[J].北方园艺,2018(10):157-160.
[14] 董慧,章炉军,张美彦,等.中国香菇主栽品种遗传多样性的SSR分析及指纹图谱构建[J].微生物学通报,2017,44(6):1427-1436.
[15] XIANG X J,LI C,LI L,et al.Genetic diversity and population structure of Chinese Lentinula edodes revealed by InDel and SSR markers[J].Mycological Progress,2016,15(4):37.
[16] 沈秀芬,章炉军,张美彦,等.利用InDel标记分析中国香菇菌株的遗传多样性与群体结构[J].菌物学报,2021,40(9):2266-2281.
[17] 张美彦,宋春艳,于海龙,等.基于SNP分型的香菇交配型AS-PCR鉴定[J].食用菌学报,2019,26(2):1-9.
[18] LI C,GONG W B,ZHANG L,et al.Association mapping reveals genetic loci associated with important agronomic traits in Lentinula edodes,Shiitake mushroom[J].Frontiers in Microbiology,2017,8:237.
[19] ZHANG J C,SHEN N,LI C H,et al.Population genomics provides insights into the genetic basis of adaptive evolution in the mushroom-forming fungus Lentinula edodes[J].Journal of Advanced Research,2022,38:91-106.
[20] 崔筱,劉芹,孔维丽,等.基于拮抗及ITS序列分析的河南香菇主产区种质资源鉴定及遗传多样性分析[J].中国瓜菜,2022,35(7):31-38.
收稿日期:2023-11-20;修回日期:2024-01-17
基金项目:河南省重点研发与推广专项(212102110056)
作者简介:宋琳琳,女,讲师,主要从事食药用真菌资源开发和分子遗传研究。E-mail:cnusll@126.com
通信作者:孔维丽,女,研究员,主要从事食用菌育种及平菇发酵料栽培机制研究。E-mail:kongweili2005@126.com
猜你喜欢
Journal of Electronic Science and Technology(2022年2期)2022-07-08 01:38:52
今日农业(2020年16期)2020-12-14 15:04:59
饮食保健(2017年15期)2017-09-03 03:35:08
中国中药杂志(2017年1期)2017-03-06 21:19:22
中外医疗(2016年33期)2017-03-02 19:04:28
中国医药导报(2015年27期)2015-10-28 20:27:29
山东体育学院学报(2015年1期)2015-06-25 20:43:38
英语学习(2015年12期)2015-02-01 14:08:30
中华皮肤科杂志(2014年3期)2014-12-19 12:54:52
特别健康·下半月(2014年8期)2014-10-28 14:20:32
