
白细胞介素-2诱导型酪氨酸激酶抑制剂研究进展
2024-03-25 09:28何思婕张男侠杨梦宇唐伟方
药学进展 2024年1期
何思婕,张男侠,杨梦宇,唐伟方
(中国药科大学理学院,江苏 南京,210009)
肝癌中表达的酪氨酸激酶(tyrosine kinase ,Tec)家族由5个成员组成:Tec、布鲁顿氏酪氨酸激酶(Bruton,s tyrosine kinase,BTK)、白细胞介素-2诱导型酪氨酸激酶(interleukin-2 inducible tyrosine kinase,ITK)、静息淋巴细胞激酶(resting lymphocyte kinase,RLK)和骨髓激酶(bone marrow X-linked kinase,BMX)。在T淋巴细胞中,有3种主要的Tec激酶表达:ITK、 RLK和Tec,它们都是在T细胞受体(T cell receptor,TCR)刺激下发生磷酸化的Tec。ITK是1992年被发现的一种具有5个结构域的蛋白激酶,其相对分子质量为72000,主要在T细胞中表达,也在肥大细胞、自然杀伤细胞(natural killer cell,NK)和自然杀伤T细胞(natural killer T cell,NKT)中表达。ITK通常位于细胞质中,激活后向质膜转移,对TCR信号传导至关重要,在辅助性T细胞1(T helper 1 cell,Th1)、Th2、Th17和恒定自然杀伤T细胞(invariant natural killer T cells,iNKT)的细胞因子产生过程中有重要作用[1-4],对非经典调节性T细胞也有调节作用[5],能调节T细胞的激活、增殖和分化。ITK通过控制促炎因子的表达参与炎症反应的发病机制,对其进行抑制是缓解或治疗急性肾损伤、多发性硬化症、银屑病、溃疡性肠炎等炎症性疾病的潜在策略。目前已有多种结构类型的小分子抑制剂被报道,其中JET-051、CPI-818分别进入Ⅱ期、Ⅰ期临床试验,但还未有ITK抑制剂上市。本文通过对ITK的结构、参与的信号通路、在炎症中的作用及其抑制剂的研究进展进行综述,以期为ITK抑制剂的开发提供参考。
1 ITK结构
ITK基因在人类染色体上定位于5q31 ~ 5q32位,由5个不同的结构域组成:pleckstrin同源(pleckstrin homology,PH)结构域、Tec同源(Tec homology,TH)结构域、肉瘤基因同源3(sarcoma homology 3,SH3)、SH2和SH1结构域(见图1)。PH结构域位于ITK的氨基端,SH1结构域位于ITK的羧基端[6]。柔性铰链连接氨基端和羧基端。连接两端的柔性铰链区形成腺嘌呤核苷三磷酸(adenosine triphosphate,ATP)结合位点的一部分,激活环位于两端激酶结构域之间[7]。ITK的PH结构域负责蛋白-脂质相互作用,并帮助激酶结合在细胞膜上。TH结构域包含27个氨基酸残基组成的保守序列,显示出对ITK家族性识别。TH结构域的脯氨酸富集区(proline rich region,PRR)与SH3结构域结合,使其处于自身抑制状态。SH3结构域包含1个保守的氨基酸残基Trp208,它对SH3结构域与PRR的结合至关重要,并导致激酶处于自抑制状态。SH2结构域负责蛋白-蛋白相互作用,使该激酶与磷脂酶Cγ1 (phospholipase C-gamma 1,PLCγ1)结合。SH2和SH3结构域的任何突变或者缺失都会导致该激酶失活。SH1结构域(激酶或催化结构域)对于激酶的催化活性,尤其是通过磷酸化该脂肪酶的Tyr775和Tyr783残基来激活PLCγ1是必要的。激酶的催化结构域包含1个ATP结合位点,将磷酸基从ATP分子转移到底物,即使底物磷酸化。该结构域还包括1个重要的氨基酸残基Tyr511,该氨基酸残基被淋巴细胞特异性蛋白酪氨酸激酶(lymphocyte specific protein tyrosine kinase,Lck)转磷酸化,用于初始激活ITK[8]。
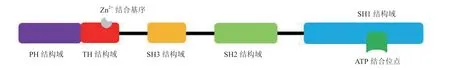
图1 ITK激酶的结构域Figure 1 The domain of ITK kinase
2 ITK在TCR信号通路中的作用
ITK是TCR介导的信号传导的关键成分。TCR与抗原提呈细胞(antigen-presenting cells,APC)上的肽-主要组织相容性复合体(major histocompatibility complex,MHC)复合物产生相互作用,从而活化Lck,导致分化簇3(cluster of differentiation,CD3)免疫受体酪氨酸活化基序(immunorecepter tyrosine-based activation motif,ITAMs)的磷酸化。接着70 kDa zeta链相关蛋白激酶(zeta chain-associated protein kinase 70 kDa,Zap-70)与磷酸化的ITAMs结合,并被Lck磷酸化,使Zap-70活化,并导致T淋巴细胞活化衔接子(linker for activation of T cells,LAT)和含有76 kDa白细胞蛋白的SH2结构域(SH2 domain containing leukocyte protein of 76 kDa,SLP-76)磷酸化。Src激酶的活化导致磷脂酰肌醇3-激酶(phosphoinositide 3-kinase,PI3K)的活化,随后生成磷脂酰肌醇(3,4,5)三磷酸(phosphatidyl-inositol-3,4,5-trisphosphate,PIP3)并在质膜上积累,进而ITK通过其PH结构域被招募到膜上,通过SH3和SH2结构域与磷酸化的SLP-76/LAT衔接复合物相互作用,并在Y511和Y180上磷酸化。ITK还与下游靶标PLCγ1相互作用并直接磷酸化,导致磷脂酶活化。PLCγ1活化后可水解磷脂酰肌醇-4,5-二磷酸(phosphatidylinositol-4,5-bisphosphate,PIP2)产生第二信使1,4,5-三磷酸肌醇(inositol triphosphate,IP3)和二酰甘油(diacylglycerol,DAG),2个第二信使都有助于传导下游信号以调节基因表达。下游信号分子包括Ca2+通道、细胞外调节蛋白激酶(extracellular regulated protein kinases,Erk)激活与转录、细胞因子释放和肌动蛋白重组(见图2)。ITK的缺失不会导致这些下游信号分子的消失,但会使它们明显减少,导致不同T细胞谱系的发育和分化发生改变[9-10]。ITK不是TCR信号传导所必须的,但它是TCR信号传导的催化剂,能够加速细胞对TCR刺激的反应速率[11]。
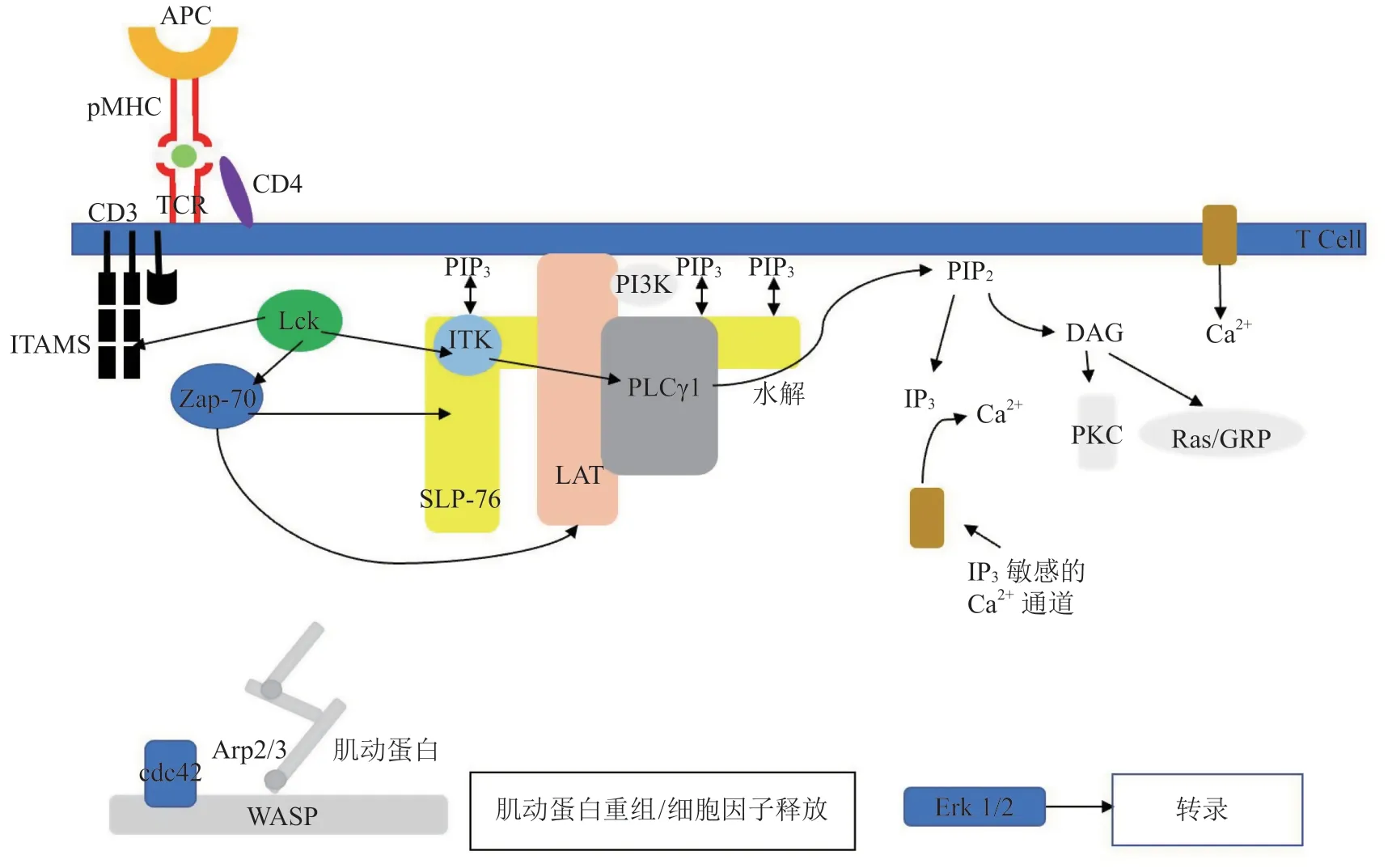
图2 ITK在T细胞信号传导中的作用Figure 2 The role of ITK in T cell signaling
3 ITK与炎症
ITK与许多炎症疾病有关。促炎性Th17细胞和抗炎性调节性T细胞(regulatory cells,Treg)之间的平衡对于产生保护性免疫应答同时最小化自身免疫至关重要,二者的失衡会导致免疫疾病的产生。ITK调节Th17和Treg免疫应答,研究表明ITK正调控Th17细胞,负调控Treg细胞[12-14]。此外,ITK负调控Th1细胞[14],在炎症中也有重要作用。
3.1 急性肾损伤
先天性或适应性免疫细胞会释放多种促炎细胞因子,它们从体循环迁移到肾组织,对肾内皮细胞和肾小管上皮细胞造成急性肾损伤(acute kidney injury,AKI)。CD4+T细胞的亚型Th1/Th17被过度激活会导致严重炎症反应,与AKI的发病有关[15-16]。CD4+T细胞中的ITK对于调节AKI过程中的炎症通路有关键作用。AKI发病过程中,ITK在T细胞中处于活化状态,抑制ITK可能导致Th1/Th17细胞因子下调和Treg细胞上调,从而使得AKI得到改善[16]。早期损伤阶段的特征是γ干扰素(interferon-γ,IFN-γ)表达上调,可能由Th1细胞产生,在后期,Th17细胞使损伤和组织纤维化持续[17]。抑制ITK可缓解AKI。
3.2 多发性硬化症
多发性硬化症(multiple sclerosis,MS)是Th1和Th17细胞侵入中枢神经系统,引起髓鞘组织损伤,导致瘫痪以及神经元损伤和丢失的一种疾病,其病变的组织病理学模式具有异质性,并且在患者之间和疾病的不同阶段有所不同[18-19]。研究表明,ITK促进CD4+T细胞迁移到中枢神经系统并导致神经炎症。ITK在抗原特异性细胞的血脑屏障迁移中可能发挥作用。因此,ITK的缺失减少了CD4+T细胞进入中枢神经系统和穿越大脑内皮屏障的转运以及Th1和Th17效应细胞因子的分泌,并且促进了Treg细胞的扩增,使神经炎症得到抑制[19]。ITK可以调节炎症T细胞进入中枢神经系统,成为MS的治疗靶点。
3.3 银屑病
银屑病是由于角质形成细胞过度增殖和皮肤中免疫细胞浸润而产生的,在患者手、脚、背部等皮肤上出现明显的银色鳞片的慢性皮肤病。研究表明,在人类皮肤上应用咪喹莫特(imiquimod,IMQ)会导致树突细胞释放白细胞介素-23(interleulcin-23,IL-23),使Th0向Th17分化,Th17分泌的细胞因子导致角质细胞功能障碍和招募中性粒细胞/树突细胞,并导致牛皮癣样炎症的发生[20]。通过使用ITK抑制剂可以阻断ITK信号通路,进而显著削弱IMQ启动的Th1/Th17免疫应答,并扩增Treg细胞,最终使银屑病得到缓解[21]。因此,抑制ITK可能是治疗银屑病型炎症的一种有效方法。
3.4 系统性红斑狼疮
系统性红斑狼疮(systemic lupus erythematosus,SLE)是一种能使多器官逐渐衰弱的慢性自身免疫性疾病,遗传、环境因素和荷尔蒙的复杂相互作用导致自身免疫耐受性崩溃,损伤皮肤、关节、肾、心脏和大脑等多个器官[22]。CD4+T细胞和CD8+T细胞是SLE发病和产生炎症的关键因素,Th17细胞和IL-17家族的细胞因子在SLE中起重要作用[23],SLE患者存在Th17/Treg细胞的相互作用,Th17细胞可能抑制Treg细胞的分化[24]。在SLE患者中表达ITK的T细胞(包括CD4+pITK+T、CD8+pITK+T细胞)增多,且与SLE的严重程度以及Th17相关细胞因子呈正相关。研究发现,表达ITK的T细胞和血清中IL-10水平之间没有显著的关联,这一初步结果不能推断ITK对Treg细胞的作用,ITK在SLE患者Treg细胞分化中的作用有待进一步探讨[25]。目前尚未有ITK抑制剂用于治疗SLE的研究。
3.5 肺炎
研究表明,急性肺损伤(acute lung injury,ALI)期间Th17细胞中磷酸化ITK(phosphorylated-ITK,p-ITK)的高表达与中性粒细胞相关炎症反应上调、肺上皮通透性受损有关。ITK通过上调Th17免疫应答和氧化应激参与脂多糖(lipopolysaccharide,LPS)诱导的肺部炎症和上皮屏障功能障碍。ITK抑制剂通过降低ALI小鼠肺室Th17免疫反应/氧化应激和上调Treg细胞,降低上皮屏障高通透性,改善气道炎症[4]。ITK阻断可能是一种减轻ALI相关气道炎症的潜在治疗策略。ITK根据肺部炎症的类型不同可以差异调节Th17细胞因子的产生,在直杆糖多孢菌诱导的Th17驱动的过敏性肺部炎症(hypersensitivity pneumonitis,HP)中,Th17细胞的IL-17A的分泌和细胞因子的产生并不需要ITK[26]。
3.6 溃疡性结肠炎
溃疡性结肠炎(ulcerative colitis,UC)是黏膜炎症从直肠开始并在结肠向近端延伸的一种慢性炎症性肠病[27]。研究结果表明,UC患者的黏膜T细胞中ITK磷酸化增加,ITK调节UC中特有的黏膜细胞因子谱,在UC中表达p-ITK的黏膜CD4+T细胞的扩增与疾病的严重程度相关[28]。选择性靶向ITK是一种治疗UC的新策略。
4 ITK抑制剂
ITK在调节致使炎症发生的信号通路中发挥核心作用。因此,将ITK作为解决T细胞相关疾病的一个关键靶点设计新抑制剂是合理的,目前已有多种结构类型的ITK抑制剂被报道(见表1)。

表1 已报道的ITK抑制剂Table 1 ITK inhibitors have been reported
4.1 氨基噻唑类
百时美施贵宝公司使用均相时间分辨荧光法(homogeneous time-resolved fluorescence,HTRF)对公司的内部化合物库进行筛选确定化合物1是一种选择性ITK抑制剂,IC50为1 μmol · L-1。通过对化合物1的噻唑环、硫甲基连接子、N-乙酰哌嗪酰胺、中心苯环、噻唑2-羧酰胺部分分别进行结构修饰,最终将硫芳基的苯环进行二甲基取代,并在苯甲酰胺的对位引入1个较大的疏水基团,优化得到BMS-488516(2),IC50为0.09 μmol · L-1。化合物2对Tec家族激酶具有较高的选择性(约500倍),在体外具有降低IL-2产生和抑制人及小鼠T细胞增殖的良好活性,但药代动力学性质不佳,在小鼠中只有高剂量(50和100 mg · kg-1)的皮下给药有一定效果[29]。因此,又在化合物1的结构基础上进行新一轮的优化,通过构效关系研究,去除了化合物1的硫甲基醚的亚甲基部分、将硫苯基替换为4-甲氧基-6-甲基类似物,并在苄胺上引入1个较大的疏水基团,最终获得了BMS-509744(3)。与化合物2相比,化合物3具有更强的ITK抑制活性(IC50= 19 nmol · L-1)和更好的药代动力学性质,但是对Tec家族激酶的选择性有所降低(约为200倍),口服生物利用度仍然较差(F= 6.4%)[30]。此外,化合物3能显著降低卵白蛋白诱导的过敏/哮喘小鼠模型的肺部炎症[31],能使心脏移植后Th1/Th17反应降低,显著延长心脏移植的平均存活时间[32],局部应用可改善小鼠银屑病样皮肤炎症[33]。

4.2 四氢吲唑类
基因泰克公司于2014年通过高通量筛选得到了化合物4,并根据化合物4与ITK的共晶结构(见图3A),对化合物4的吲哚的6位、吡唑杂环、氰苯基取代以及苄位取代进行改造,得到了一系列对ITK表现出较强的抑制活性(Ki<1 nmol · L-1)和良好的激酶选择性的化合物,其中化合物5的Ki为0.1 nmol · L-1,对磷脂酶Cγ(phospholipase Cγ,PLCγ)的 IC50为25 nmol · L-1。可能由于渗透性较低,与化合物4相比,其清除率更低,且没有表现出良好的生物利用度[34]。为此,在后续的优化中将同时改善化合物的溶解度和渗透性。在第一轮的优化中,用四氢吲唑替代化合物4的吲哚母核以降低芳环数量,从而降低溶解度预测指数(solubility forecast index,SFI),以期改善溶解度。在四氢吲哚母核上引入了不同的取代基后发现,引入偕二甲基能增强对ITK的抑制活性(Ki= 5.3 nmol · L-1),但溶解度并未得到提升。在第二轮的优化中,进一步在苯环苄位引入与化合物5相同的碱性增溶基团,得到四氢吲唑类抑制剂GNE-9822(6)。共晶结构显示,化合物6的四氢吲唑母核上的偕二甲基伸入邻近Phe435的亲脂性口袋,在铰链区形成3个氢键,并以苄基为中心使苯基与Phe437形成合适的π-π相互作用(见图3B)。该化合物具有良好的溶解度和中等的渗透性,在人肝细胞中有较低的清除率和较高的人血浆蛋白结合能力,在动物中有良好的生物利用度和较长的半衰期[35]。但在进行体内疗效研究时发现其在肝细胞和Jurkat细胞中具有脱靶效应和细胞毒性。为消除这些缺点,笔者对已合成的四氢吲唑类抑制剂进行分析,发现细胞毒性与化合物的碱性相关。为此,通过用烷基砜代替碱性胺来降低细胞毒性,用二氟亚甲基环丙基取代化合物6的偕二甲基以优化配体与Phe435附近亲脂口袋之间的相互作用来提高抑制作用,最终开发了GNE-4997(7),其结合模式与化合物6的结合模式一致。酰胺NH与Met438的羰基结合,吡唑与Met438的NH和Glu436的羰基形成氢键作用,从而将抑制剂固定在铰链区。环砜用于固定苯环与Phe437的π-π相互作用。环丙烷的二氟亚甲基位于相邻Phe435的亲脂口袋中(见图3C)。化合物7的Ki值为0.09 nmol · L-1,对Jurkat细胞PLC-γ磷酸化的抑制作用IC50为4 nmol · L-1,对Jurkat 细胞增殖的IC50为12 μmol · L-1,与化合物6相比,其对Lck的选择性增加,肝毒性也有所降低[36]。
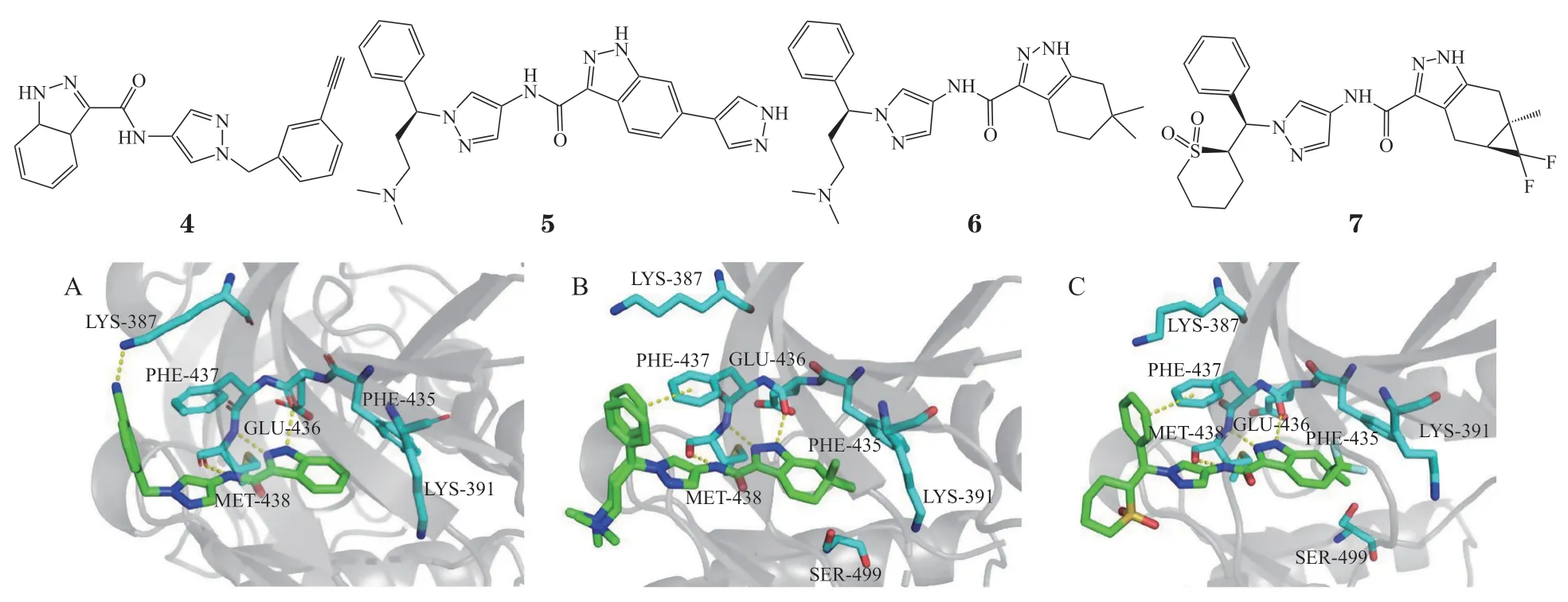
图3 化合物4、6、7与ITK激酶结合模式图Figure 3 The binding pattern of compound 4、6、7 with ITK
4.3 苯并咪唑类
异常活化的T细胞或NK细胞与许多疾病高度相关。由于在T细胞的发育过程中,RLK和ITK共同调节TCR信号通路[37],因此开发ITK/RLK双靶抑制剂将有利于有效阻断Tec激酶驱动的这些细胞的活化。2015年,Zhong等[38]通过共价靶向技术和基于结构的设计发现了高选择性的苯并咪唑类ITK/RLK抑制剂PRN694(8)。化合物8通过与Cys442ITK或Cys350RLK形成一个共价键(见图4),不可逆地抑制ITK和RLK,IC50分别为0.3和1.4 nmol · L-1。化合物8可阻止TCR或Fc受体(FcR)诱导的细胞活化,抑制TCR诱导的T细胞增殖而不产生直接的细胞毒性,并阻断促炎细胞因子的释放。在噁唑酮诱导的迟发性超敏反应(delayed type hypersensitivity,DTH)小鼠模型中,化合物8能减轻迟发性超敏反应[38]。此外,化合物8对Th1分化、IFN-γ的产生、Th17分化和IL-17A的产生具有较强的抑制作用,而对Th2分化的抑制作用较弱[39]。研究表明,化合物8在Rac1v12小鼠中能抑制T淋巴细胞功能,降低全身肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)含量,减少促炎细胞因子释放,并且在IMQ小鼠模型中对银屑病有一定的治疗效果,能够有效降低银屑病样表型的严重程度,并减少表皮增殖和厚度[40]。这些研究结果表明, ITK/RLK双靶点抑制剂在治疗T细胞或NK细胞恶性肿瘤以及炎症和自身免疫性疾病,如银屑病、类风湿关节炎、MS和炎症性肠病方面具有良好的应用前景。

图4 化合物8与ITK活性部位对接图(PDB ID: 3QGY)Figure 4 Molecular docking model of compound 8 with ITK active site(PDB ID: 3QGY)
4.4 吡唑并嘧啶类
吡唑并嘧啶类化合物ibrutinib(9)是BTK的不可逆抑制剂,ITK与BTK具有显著的序列同源性和功能相似性,并且都包含ibrutinib结合位点,该结合位点由1个SH3自磷酸化酪氨酸(tyrosine,Tyr)和1个共价结合半胱氨酸残基组成。BTK和ITK之间的同源性,以及ibrutinib在ITK Cys442处的潜在共价结合(见图5)和活性位点的占有率与ibrutinib不可逆结合BTK时的情况类似,使ibrutinib具有ITK活性,IC50为2.2 nmol · L-1。分析证实ibrutinib不可逆地结合ITK并抑制TCR刺激后Th2细胞的活化。由于RLK同样可以使Th1和CD8+T细胞的活化,因此这种抑制对Th2极化CD4+T细胞具有特异性[41]。此外,Ibrutinib阻断人Th17细胞和相关细胞因子的体外生成并提高叉头框蛋白P3(forkhead box protein P3,FOXP3)表达。低剂量ibrutinib可改善人CD4+T细胞体外分化为Treg细胞,高剂量的ibrutinib完全阻断了Treg细胞的产生[42]。实验结果证实,Ibrutinib是首个具有临床可行性的ITK强效和不可逆抑制剂。
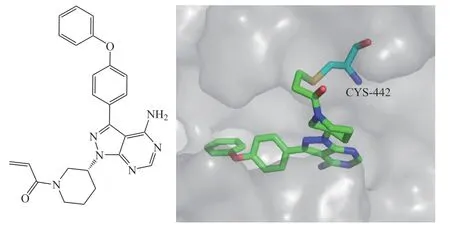
图5 化合物9与ITK活性部位对接图(PDB ID: 3QGW)Figure 5 Molecular docking model of compound 9 with ITK active site(PDB ID: 3QGW)
PF-06465469(10)是辉瑞公司研发的吡唑并嘧啶类共价抑制剂。它是以化合物11为基础进行改造。化合物11是通过化合物库筛选得到的共价激酶抑制剂,对ITK的IC50为73 nmol · L-1。对化合物11进行细胞肌醇单磷酸酶(inositol monophosphatase,IP1)测试,与酶水平测试结果相比,化合物11在细胞中的效果显著下降,IC50为2.21 μmol · L-1。为替换不利于代谢的叔丁基,Zapf等[43]设计合成了一系列苯胺、烷基胺衍生物,其中异丙基苯基衍生物(10)的活性有显著提高,对ITK的IC50为2 nmol · L-1,在细胞和人全血(human whole blood,hWB)分析中也有较好的作用效果,IC50分别为31和48 nmol · L-1。化合物10与ITK蛋白的对接结果显示,丙烯酰胺与Cys442形成共价相互作用,2个芳基之间的酰胺键与Lys391和Asp500形成氢键作用(见图6)。蛋白结合动力学模拟实验结果表明化合物10对ITK具有良好的亲和力,并且不可逆地与ITK结合,但其对ITK和BTK 2种激酶具有相当的抑制活性,这表明仍需要通过适当的结构修饰,来实现选择性的提升[43]。

图6 化合物10与ITK活性部位对接图(PDB ID: 4HCU)Figure 6 Molecular docking model of compound 10 with ITK active site(PDB ID: 4HCU)
4.5 吡啶酮类
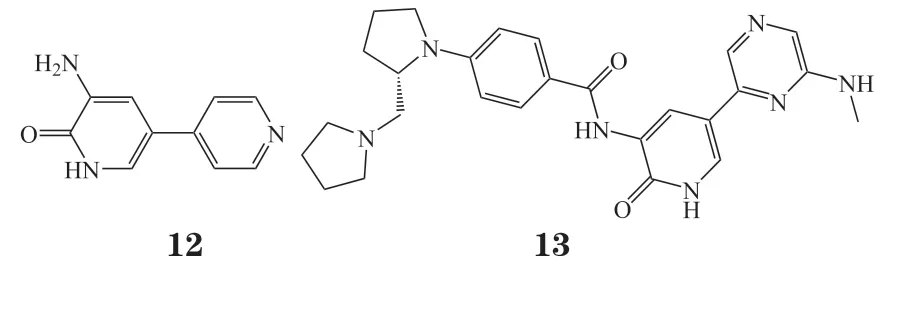
由于3-氨基吡啶-2-酮结构具有与激酶形成多个氢键的潜在能力,因此Charrier等[44]选择该片段作为核心骨架,以化合物Amrinone(12)为设计起点,用苯甲酰基替代氨基,并在苯甲酰胺环上引入亲脂性基团,对吡啶环进行结构修饰,并使用带有SP评分的Glide对接软件进行虚拟筛选得到吡咯烷甲基吡咯烷基团以增加溶解度,设计了一系列吡啶酮类化合物,其中化合物13对ITK显示出良好的抑制活性(Ki= 7 nmol · L-1)与BMS-509744相当,具有良好的选择性。该化合物具有符合口服类药物特征的理化性质,在pH = 7.4时溶解度为150 μg · mL-1,渗透性良好,外排较少,在渗透性实验Caco2的研究中,A-B为10.8×10-6cm · s-1,B-A为32.4×10-6cm · s-1。
4.6 磺酰吡啶类
Trani等[45]通过对基因泰克公司高通量筛选得到的化合物14和葛兰素史克公司发布的ITK抑制剂化合物15的结构分析,进一步改造得到了化合物16。与化合物15相比,化合物16具有更好的水溶性,但化合物16对Lck缺乏选择性,且在Jurkat细胞的测试中活性较差(IC50= 2.6 μmol · L-1)。为此,Trani等[45]用苯磺酰基替代苄基,用吡啶环替代嘧啶环来最大化苯环与ITK的Phe437的π-π相互作用以提高对Lck的选择性,并在铰链区用不同的基团进行取代,设计并合成了一系列磺酰吡啶类化合物。其中化合物17的细胞活性(0.17 nmol · L-1)和对Lck的选择性(182倍)较好,这是因为其环戊烷环在ITK中与Phe435能形成更强的亲脂性相互作用。该化合物氨基吡唑部分与铰链序列(Met438和Glu436)形成3个氢键,砜连接子使苯环与吡啶环形成了60°夹角,从而形成了π-π相互作用,环己醇的羟基与水分子形成氢键,并通过与Ser499和Asp500形成氢键而更加稳定(见图7)。化合物17能够抑制Jurkat细胞的PLC-γ1磷酸化(IC50= 180 nmol · L-1),在人肝微粒体中表现出中高的清除率,在MDR1/MDCK的试验中表现出中等的渗透性,但它的溶解度等理化性质仍需要进一步改善。

图7 化合物17与ITK活性部位对接图(PDB ID: 4QD6)Figure 7 Molecular docking model of compound 17 with ITK active site(PDB ID: 4QD6)
4.7 吲哚基吲唑类
以化合物18(ITK IC50= 10.9 nmol · L-1)为先导化合物,Wang等[46]用不同的亲电弹头修饰烷氧基醚侧链与Cys442形成共价键,以及在吲唑的6位引入不同的取代基占据Phe435、Lys391、Val377和Ala 389形成的疏水口袋以提高其活性,在吲哚的5位引入不同的芳基取代与Phe437形成T型π-π相互作用以提高对BTK的选择性,合成了一系列吲哚啉唑类ITK抑制剂。对接结果显示,化合物19(ITK IC50= 4 nmol · L-1)与ITK形成了3个氢键,其苯基近似垂直于Phe473的苯环(见图8)。试验结果表明,化合物19和20(ITK IC50= 5.8 nmol · L-1)可以有效抑制ITK下游底物的磷酸化,当浓度为0.3 μmol · L-1时,Jurkat细胞中的PLCγ1磷酸化被完全抑制(化合物19,EC50= 119 nmol · L-1;化合物20,EC50= 133 nmol · L-1),此外,也抑制Jurkat细胞中ERK1/2的磷酸化,在细胞中对BTK表现出优异的选择性。化合物19和20具有一定的抗增殖活性,在低微摩尔水平上抑制3种T-ALL细胞(Jurkat,MOLT-4和 CCRF-CEM细胞)和一种皮肤T细胞淋巴瘤细胞(H9)的生长,但对HEK 293 T细胞的抑制作用较弱。化合物19的半衰期较短、清除率高、分布体积小,因此其可能残留在循环系统中。
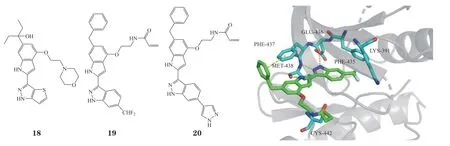
图8 化合物19与ITK的ATP结合口袋对接图(PDB ID:3V5J)Figure 8 Molecular docking model of compound 19 with ATP binding pocket of ITK (PDB ID: 3V5J)
4.8 吡咯并嘧啶类
化合物21是表皮生长因子受体T790M突变体的不可逆抑制剂,在1 μmol · L-1时对ITK的抑制率超过80%,通过参考化合物21的结构,Tang等[47]以7H-吡咯并[2,3-d]嘧啶骨架为起点对该分子进行合理的设计和改造,进一步提高了其选择性,从而获得针对ITK的选择性共价抑制剂。实验结果表明,化合物22表现出对ITK的有效活性(IC50= 3.7 nmol · L-1)和对BTK的优异选择性。化合物22还可以抑制Jurkat细胞中ITK对PLCγ1的磷酸化作用,并对几种T细胞白血病和淋巴瘤细胞系显示出良好的抗增殖作用。

4.9 未公开结构
JTE-051和CPI-818是2个已进入临床试验阶段但结构尚未公开的ITK抑制剂,其基本信息见表2。

表2 临床试验中的ITK抑制剂Table 2 ITK inhibitors in clinical trials
JTE-051是日本烟草株式会社研发的目前唯一进入临床试验阶段,用于治疗类风湿关节炎和银屑病的ITK抑制剂,通过抑制激活与免疫反应相关的T细胞的信号来抑制过度活跃的免疫反应,其中治疗类风湿关节炎(NCT02919475)、牛皮癣(NCT03358290)、神经性疼痛(jRCT2031210042)已进入II期临床试验。 2021年6月公开的治疗类风湿关节炎的II期临床数据表明[48],JTE-051在不同的给药剂量(50、100、150和200 mg)下均具有一定的疗效,在258名受试者中仅有8名出现严重不良反应,表现为胃肠道功能紊乱(2人)、感染和侵袭(3人)、肌肉骨骼和结缔组织疾病(3人)。
CPI-818是Corvus 制药研发的一种口服的ITK共价抑制剂,其IC50为2.3 nmol · L-1,选择性比RLK和BTK高100倍,能抑制外周血单核细胞(peripheral blood mononuclear cell,PBMC)中ERK和PLCγ的磷酸化,并抑制Jurkat细胞中IL-2的分泌(IC50= 75 nmol · L-1)[49],可以有效抑制TCR信号的转导,且对双阴性T细胞(double negative T cell,DNT)没有毒性[50]。在自身免疫性淋巴增生综合征(autoimmune lymphoproliferative syndrome,ALPS)的MRL/lpr小鼠模型中,CPI-818能显著降低DNT细胞数量、抑制淋巴结病变和其他疾病的发生。此外,研究数据表明,针对ITK阻断TCR信号传导可能是治疗成人和儿童ALPS的一种有效策略[51]。临床相关移植物抗宿主病(graft versus host diseases,GVHD)模型研究数据表明,抑制ITK可通过抑制T细胞的激活和增殖、降低促炎细胞因子的浓度和增加抗炎因子的浓度来预防急性移植物抗宿主病(acute graft versus host disease,aGVAD)[52]。CPI-818在T细胞淋巴瘤的Ⅰ/Ⅰb期临床试验(NCT03952078)中的评估结果显示其具有良好的耐受性和抗肿瘤活性,但其本身的化学结构、优化过程尚未公开。Ⅰ期临床数据表明CPI-818通过保留RLK,选择性抑制ITK,诱导Th1细胞偏斜,以剂量依赖的方式影响T细胞的分化、增殖,在剂量递增期间未观察到剂量限制性毒性(dose limited toxicity,DLT),接受200 mg剂量治疗的患者达到的药物血浆浓度与体外观察到的Th1偏斜一致,该剂量下的6名外周T细胞淋巴瘤患者中有1名维持了15个月的完全缓解(complete remission,CR),有2名病情稳定(stable disease,SD)[53]。
5 结语与展望
ITK在T细胞信号传导、分化、发育和细胞因子产生中发挥重要作用,通过调控促炎细胞因子的表达,能够影响免疫性炎症的发生、发展。目前研究人员虽然设计合成了多种结构的ITK抑制剂,但由于选择性不佳、理化性质不佳、药代动力学性质不佳等原因,仍没有药物上市,只有JTE-051进入Ⅱ期临床。由于ITK与BTK之间显著的同源性,BTK抑制剂如ibrutinib,成为了临床可行的ITK抑制剂。ITK与RLK的协同作用使得ITK/RLK双重抑制剂能产生更有效的抗炎作用。随着各个化合物与ITK的共晶结构被报道,人们对其了解更加深入,相信终将开发更安全有效的ITK抑制剂,使患者获益。
猜你喜欢
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
天津医科大学学报(2019年6期)2019-08-13
广州大学学报(自然科学版)(2019年1期)2019-05-07
分析化学(2017年12期)2017-12-25
天津科技大学学报(2016年1期)2016-02-28
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10
中国医药生物技术(2015年4期)2015-12-26
安徽医科大学学报(2015年9期)2015-12-16
现代检验医学杂志(2015年2期)2015-02-06
