
转甲状腺素蛋白抑制β淀粉样蛋白聚集的分子机制研究*
2024-03-23 07:10周双艳黄垚心白佳慧
生物化学与生物物理进展 2024年3期
周双艳 黄垚心 李 鑫 白佳慧 袁 帅
(重庆邮电大学生物信息学院,大数据生物智能重庆市重点实验室,重庆 400065)
阿尔茨海默病(Alzheimer’s disease,AD),俗称老年痴呆症,是一种典型的神经退行性疾病,主要病理特征是脑萎缩和脑细胞死亡。该病自1906 年被首次报道以来已有百年之久[1],但到目前为止有关AD 的发病原因仍未明确,这给AD 的治疗及药物研发带来了巨大的挑战,有上百种药物在研发阶段纷纷宣告失败。AD药物研发的困难很大程度上可归结为极为复杂的AD发病机制。截至目前,学术界为解释AD的发病机制提出了诸多假说,如β 淀粉样蛋白(amyloid beta protein,Aβ)级联学说、Tau 蛋白异常磷酸化学说、胆碱能学说、神经炎症等[2]。尽管这些学说的聚焦点有所差异,但由此也说明AD的发病可能为多因素协同致病。此外,有研究表明,一些周边及全身性的异常与AD 有关,并推测AD 不止是一种脑部疾病,而是一种全身性的疾病[3]。AD复杂的病因无疑加重了治疗药物研发困难。围绕AD发病机制的各种学说,目前抗AD药物研发的主流策略包括清除沉积的β淀粉样斑块[4]、调节胆碱能系统[5]、减少炎症反应和氧化损伤[6]以及多靶点治疗策略[7]等。然而,近些年来在这些策略的指导下,仅有Aducanumab、Lecanemab 被美国食品药品监督管理局(FDA)批准上市[8-9]。这两款药物均由渤健和卫材公司联合开发,是通过清除AD患者大脑内β淀粉样斑块而发挥作用的单克隆抗体药物。面对AD药物研发的严峻形式和挑战,如何从困境中寻找新的突破点仍然是AD药物研发的重要方向。有研究表明,人体内转甲状腺素蛋白(transthyretin,TTR)对AD具有神经保护作用,这种神经保护作用能够延缓AD的疾病进程[10-12]。这一发现在一定程度上可为AD的药物设计提供新的思路。
TTR是一种转运蛋白,负责甲状腺素和视黄醇在人体内的转运工作[13]。正常情况下,TTR 是一种同源四聚体蛋白,其四聚体结构非常稳定。但当TTR四聚体解离成单体后,TTR单体会迅速发生错误折叠并聚集形成淀粉样纤维,最终导致TTR 淀粉样变性,如淀粉样心肌症。尽管TTR 的错误折叠与TTR淀粉样变性密切相关,但多项证据表明,TTR对AD具有神经保护作用。一方面,与健康同龄人相比,AD患者脑脊液及血浆中TTR含量明显降低[14-16],且脑脊液中TTR水平与AD的严重程度和老年斑的含量呈负相关[17]。此外,Buxbaum等[18]报道,在APP23 转基因AD 小鼠模型中过表达人类野生型TTR基因能够抑制小鼠模型AD的疾病进程。这些研究结果均证明TTR直接参与了AD的发病过程。另一方面,已有研究表明,TTR 对AD 的这种神经保护作用主要体现在TTR 能够与Aβ 发生相互作用并抑制Aβ 的聚集和细胞毒性[19-20]。Aβ 是一种AD 相关蛋白,其毒性聚集体是AD 患者大脑中淀粉样斑块的主要成分。鉴于TTR 与Aβ 之间相互作用对AD 的神经保护作用,研究者们纷纷开展了相关的研究。
近期,Cotrina等[21]提出了一种靶向TTR的抗AD药物研发策略。他们通过计算机药物再利用和体外生物分析方法筛选了一组能够作为分子伴侣增强TTR/Aβ 相互作用的小分子化合物,其中3 个作为药物再利用的上市药能够直接进入临床阶段成为AD 候选药物。Cotrina 等[22]也通过量热研究确定了一个能够增强TTR/Aβ 相互作用的小分子伴侣。除小分子伴侣的策略外,Tonali 等[23]提出将蛋白质水解靶向嵌合体(proteolysis targeting chimera,PROTAC)方法用来治疗AD。该方法是一种多功能多靶点治疗策略,其原理同样是靶向增强TTR/Aβ 之间蛋白质-蛋白质相互作用从而更好地发挥TTR的神经保护作用。这些研究都表明,增强TTR与Aβ 之间的相互作用可作为潜在的抗AD 药物研发策略。
在TTR/Aβ 作用机制研究方面,Du 等[24]研究表明,TTR 单体是Aβ 单体的主要结合对象,而TTR四聚体则更易与Aβ聚集体结合。Saelices课题组[19]最近的一项研究则表明,TTR四聚体主要通过与Aβ 单体结合的方式将单体“扣押”来达到抑制Aβ聚集的效果,而只有解离后的TTR单体才能与Aβ 的低聚体结合并诱导其进一步形成更高聚合度的无毒聚集体。这一结论与此前Garai 等[25]的报道一致。与Saelices 等的结论不同,Ghadami等[26]研究表明,TTR 四聚体和单体都能与Aβ 低聚体结合,二者主要通过抑制Aβ 的初级成核和二级成核过程来抑制Aβ 低聚体的毒性和纤维增长能力。因此,到目前有关TTR与Aβ发生相互作用的结构形式仍没有确定性的结论。此外,在TTR 与Aβ 相互作用过程中,TTR105-117、TTR38-42以及TTR结构中的β折叠链A、EF螺旋片段等是目前报道的能够与Aβ发生相互作用的重要片段[27-29]。
上述研究在一定程度上提供了TTR与Aβ 相互作用的线索,但二者作用的详细机制仍然未知,且现有研究以宏观实验发现为主,微观层面上Aβ 与TTR 作用的结构动力学及热力学信息不足。基于此,本工作拟采用蛋白质-蛋白质对接及分子动力学模拟方法从分子水平上来探究TTR与Aβ的相互作用。相比宏观实验技术,分子动力学模拟能提供原子分辨率上二者相互作用过程中蛋白质结构随时间变化的动力学信息,以期从微观角度阐释TTR与Aβ相互作用的分子机制,从而为基于TTR神经保护作用机制的抗AD药物设计提供理论依据。
1 材料与方法
1.1 结构准备
Aβ 聚集体的结构从PDB 结构数据库中获得(PDB ⅠD:5OQV[31])。该结构是一个通过冷冻电镜技术辅以固体NMR 实验获得的近原子分辨率的纤维聚集体结构,结构中所有42 个残基的骨架和几乎所有的侧链在冷冻电镜密度图中都得到很好的解析。因此,该结构能够提供相对完整的Aβ 聚集体的结构信息。研究表明,较小的可溶性Aβ 低聚体是Aβ细胞毒性的主要形式[32],因此本工作以五聚体作为Aβ低聚体的代表形态进行研究(图1b)。此外,PDB ⅠD 为1F41 的晶体结构[33]被用于构建TTR四聚体及单体的结构,该晶体结构为1.30 Å分辨率下的野生型TTR 二聚体三维结构。因此,TTR 单体通过提取1F41 结构中的单体坐标获得,而TTR 四聚体则通过在VMD 中执行1F41 的旋转矩阵获得。TTR 单体及四聚体结构分别如图1c 和图1d所示。
1.2 蛋白质-蛋白质对接
为了获得用于描述TTR/Aβ相互作用的复合物结构,进行蛋白质-蛋白质的分子对接。在蛋白质-蛋白质对接过程中,本文采用了HawkDock在线网站(http://cadd.zju.edu.cn/hawkdock/)。HawkDock是侯廷军教授课题组开发的用于蛋白质-蛋白质对接的在线网站,能够实现1 000个氨基酸以内的蛋白质-蛋白质对接并预测蛋白质作用的关键残基[34]。为探究TTR及Aβ不同结构形态对二者相互作用的影响,分别对TTR单体/Aβ单体、TTR四聚体/Aβ单体、TTR单体/Aβ五聚体及TTR四聚体/Aβ五聚体进行了分子对接,各体系分别标记为AβTTRM、 AβTTRT、 AβOTTRM 和AβOTTRT。由于正常情况下Aβ 单体为非结构多肽,具有形态多样的特点。因此,为考虑Aβ 单体构象的影响,首先对Aβ 进行200 ns 的分子动力学模拟,并对模拟轨迹进行聚类分析,提取聚类含量最高的三类作为初始构象进行分子对接(图1a),对接后选取各构象打分最高的作为对接复合物的代表构象。针对Aβ 五聚体体系则分别选用打分较高的前三类作为复合物的代表构象。各体系对接结果如图2所示。
1.3 分子动力学模拟
为了深入探究TTR与Aβ的相互作用过程,进一步对各体系进行了分子动力学模拟,用于模拟的初始结构为上述分子对接所得的各体系的复合物结构。在模拟文件准备中,AMBER FF14SB 力场[35]用于描述蛋白质的原子作用。为描述溶剂效应,在蛋白质周围增加距蛋白质边缘12 Å 厚度的方形水盒子,水分子用TⅠP3P 水模型[36]描述。随后,对准备好的复合物结构先进行了10 000步的能量最小化,用于消除初始结构中不合理的原子接触。之后,对体系进行200 ps的升温模拟,使体系温度升至310 K。当体系升温至目的温度后,在NPT系综下对体系进行500 ps的平衡模拟使得体系各项参数(密度、能量等)达到平衡。最后,对平衡后的体系在NPT系综下进行200 ns的轨迹动力学模拟。模拟过程中的温度通过Langevin方法控制,压强通过Langevin Nosé-Hoover方法控制。SHAKE算法用于限制所有含氢键的键长,非键相互作用截断值设为10 Å,PME 算法用于计算长程静电相互作用。模拟的积分步长为2 fs,模拟过程中每2 000 步(4 ps)输出一个结构,200 ns共输出50 000个构象用于轨迹分析。所有模拟均使用NAMD 2.13 软件完成[37]。
1.4 结合自由能计算
MM-GBSA 方法[38-40]用于评估各体系模拟过程中TTR与Aβ相互作用的结合亲和力。该方法在计算结合自由能时将溶剂视为均匀的连续介质,并基于力场和隐式的连续介质模型对平衡轨迹结构进行平均。基于此,选用平衡轨迹的最后20 ns 进行MM-GBSA 计算,计算过程中每100 ps 提取1 个快照,共提取了200 个快照结构用于评估TTR 与Aβ相互作用的强弱。MM-GBSA计算的公式如下:
其中,ΔEvdw和ΔEele分别表示气相中的范德华能量项及静电能量项。这两项通过在气相状态下的AMBER FF14SB力场下计算获得。ΔGpolor和ΔGnonpolor分别代表极性溶剂化能和非极性溶剂化能。极性溶剂化能量项ΔGpolor通过Generalized Born(GB)模型计算获得,计算过程中溶质和溶剂的介电常数分别设为1 和80。非极性溶剂能量项ΔGnonpolor通过计算溶剂可极表面积(solvent accessible surface area,SASA)获得,计算公式为ΔGnonpolor=γ×SASA。在进行SASA 计算时,水分子的探针设置为1.4 Å,表面张力常数γ为0.007 2 kcal/(mol·Å2)。随后,进一步将结合自由能分解到每个残基上以此来确定TTR 与Aβ 相互作用过程中的重要残基。MMGBSA计算所用软件为AMBER 18[41]。
2 结果
2.1 TTR与Aβ相互作用模式分析
研究表明,TTR 及Aβ 的结构形态能够影响二者的相互作用。基于这一依据,通过分别对不同结构形态的TTR及Aβ进行分子对接以探究二者的相互作用模式,包括TTR 的单体及四聚体,Aβ 单体及低聚体(五聚体)。对接获得的复合物结构如图2示,图中分别展示了根据MM-GBSA方法的对接结合自由能以及参与形成蛋白质界面氢键的残基。
从图2 中可以看出TTR 单体与3 个不同Aβ 单体构象的结合自由能整体较高,分别为-35.06 kcal/mol、-37.52 kcal/mol以及-38.54 kcal/mol(均为各Aβ单体构象打分最高的结合模式)。其中,AβTTRM 体系的Model1复合物结构中Aβ 单体结合在TTR 单体的F 折叠链,Model2复合物结构中Aβ 单体主要结合在TTR 单体的DAGH β 折叠片层界面上,该界面在TTR 四聚体中被包裹在内部,是甲状腺素(thyroxine,T4)疏水结合通道的组成部分,Model3复合物结构中Aβ 主要结合在TTR单体EF 螺旋(图1c) 及其周围残基位置。AβTTRT体系中第一个构象与TTR四聚体的结合自由能打分最高,为-32.33 kcal/mol,这一结合模式中Aβ单体结合在TTR四聚体的T4结合位点的通道中。AβTTRT体系其他两个构象在TTR四聚体上的结合位点大致相同,大致位于T4 结合通道侧面的凹槽部位,与TTR 四聚体结构中的视黄醇结合蛋白结合位点接近[42],这一位点包括EF 螺旋loop区、AB loop 区、GH loop 区等。并且,视黄醇结合蛋白也曾被报道能够参与TTR 抑制Aβ 聚集的过程[43]。
在Aβ 五聚体与TTR单体及四聚体的对接体系中,复合物的代表构象为打分较高的前3个结合模式,可以看出AβOTTRM 及AβOTTRT 体系的Model3亲和力均较低,分别为-6.72 kcal/mol 和-1.85 kcal/mol,表明这两个模式均不利于TTR 与Aβ 五聚体的结合。有趣的是,在AβOTTRM 体系中Model1的结合亲和力非常高,为-48.47 kcal/mol,在这一结合模式中TTR 单体的β 折叠链H 与Aβ 五聚体的β折叠边链之间形成了较多的氢键,包括骨架氢键和侧链氢键,这些氢键的形成能够在一定程度上稳定TTR 单体与Aβ 五聚体的作用,尤其是TTR 单体与Aβ 五聚体β 折叠链之间的骨架氢键,表明TTR单体具有参与Aβ聚集并形成更高聚合度复合物的潜力。值得一提的是,两个TTR单体的H链通过骨架氢键形成具有分子间β折叠片层的TTR二聚体[44],TTR 二聚体进一步组合形成了TTR 四聚体。这表明TTR单体中的H链是较好的β折叠链结合部位,与本工作的结果相符。AβOTTRM体系的Model2中TTR单体结合在Aβ五聚体β片层的平面之上,参与作用的残基主要为带电氨基酸,包括Arg(R)、Asp(D)和Glu(E)。表明静电作用力是该模式的重要作用力类型。AβOTTRT 体系中Model1的结合亲和力为-36.28 kcal/mol,主要作用界面为Aβ五聚体β折叠边链与TTR四聚体T4结合位点通道的侧面凹槽,这与AβTTRT 体系的Model2及Model3中的作用位点大致相同。
2.2 TTR与Aβ相互作用的动力学模拟
在上述部分,通过蛋白质-蛋白质分子对接的方法初步探究了不同结构形态的TTR及Aβ之间可能的相互作用模式。但在这些模式中,TTR是否能够与Aβ 持续稳定地发生作用以及作用过程中是否对二者的结构产生影响等仍不清楚。为了探究TTR与Aβ 相互作用的动态过程,本文进一步对各结合模式分别进行了200 ns的分子动力学模拟。同时以Aβ 的结构为参照,对Aβ 单体及Aβ 五聚体各进行了200 ns 的动力学模拟,分别记为AβM 和AβO。这里,Aβ 单体的模拟构象为图1a 中最左边的构象,即聚类分析含量最大的构象进行模拟。
杨译:...whoever knows the truth can be a teacher.[5]151
首先通过计算各体系Aβ单体和五聚体Cα原子的均方根偏差 (root-mean-square deviation,RMSD)来监测模拟过程中Aβ 结构的稳定性,计算过程中选用各模拟轨迹的第一帧作为参考结构。AβM体系在所有Aβ单体结合的体系中的RMSD值最大(图3a),表明TTR 单体及TTR 四聚体与Aβ单体结合后,在一定程度上限制了Aβ的构象变化,使得其更好地维持原有构象。除AβOTTRT2 体系在最后40 ns 经历了较大的RMSD 波动外,其他体系的RMSD 曲线接近或低于AβO 体系,基本维持在3 Å 以下(图3b)。这表明除AβOTTRT2 体系,其他模式下Aβ聚集体与TTR单体和四聚体作用能够较好地保持Aβ 聚集体的结构,即作用过程中TTR 单体及四聚体对Aβ 聚集体整体结构的影响较小。

Fig. 3 Time evolution of the RMSD of Cα atoms for each simulated system
随后,为了评估各体系在动态模拟过程中不同结构形态的TTR 是否能够与Aβ 稳定作用,利用MM-GBSA 方法分别计算各体系TTR 与Aβ 之间的结合自由能(表1)。从表1可以看出在动态相互作用过程中不同构象的Aβ单体与TTR单体和四聚体的结合自由能较高,且大部分结合模式相较于蛋白质-蛋白质对接中的打分自由能有不同程度的增强,尤其是AβTTRT体系中3个结合模式的结合自由能增加显著, 分别由对接打分过程中的-32.33 kcal/mol、-18.99 kcal/mol以及-24.19 kcal/mol增 加 到 -43.45 kcal/mol、 -38.47 kcal/mol 和-32.73 kcal/mol。这一结果表明TTR四聚体在动态作用过程中能够通过动态结构调整增强其与Aβ 单体的作用,同时也说明TTR 四聚体具有较强的“扣押”Aβ单体的潜力。
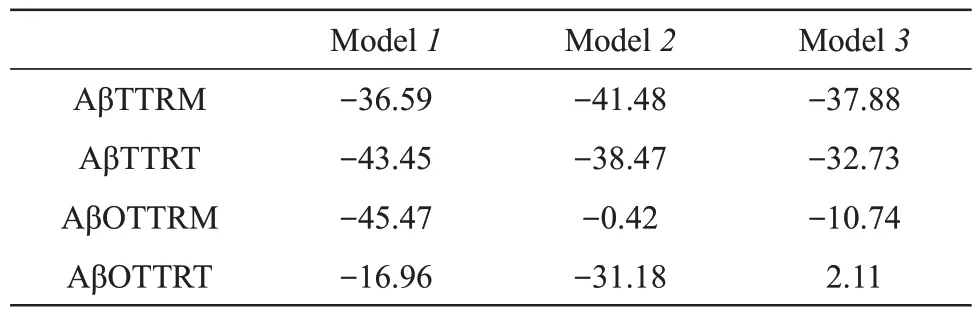
Table 1 Binding free energy for interactions between different structural forms of TTR and Aβ(energy unit: kcal/mol)
对比Aβ 单体体系,在Aβ 低聚体体系中除AβOTTRM 体系的Model1和AβOTTRT 体系中的Model2有较高的结合自由能外,其他体系的结合自由能均较低,尤其是AβOTTRM 体系的Model2和AβOTTRT 体系的Model3,结合自由能分别为-0.42 kcal/mol和2.11 kcal/mol,表明这两个作用模式不能持续稳定地结合。此外,将Aβ 低聚体体系与蛋白质-蛋白质对接过程中的结合自由能打分相比,仅AβOTTRM 体系的Model1保持了较高的结合强度,为-45.47 kcal/mol,而在AβOTTRT 体系中原来打分较高的Model1在动态作用过程中结合自由能强度降低至-16.96 kcal/mol,但原来打分较低的Model2结合自由能由原来的-10.45 kcal/mol增加至-31.18 kcal/mol。从图3b的RMSD曲线中可以看出在最后40 ns,Model2结合模式中的Aβ 五聚体经历了较大的结构变化。因此,推测AβOTTRT Model2结合自由能的增加与Aβ 五聚体构象调整有关。
随后,为了确定TTR与Aβ动态作用过程中对二者相互作用起关键作用的关键残基,本文对各体系结合自由能最高的结合模式进行了残基能量分解,即将作用过程中总的结合自由能分解到各个残基中。同时,为了更直观地观察模拟后的结构特征,将上述模式的动力学轨迹进行了聚类分析,并提取聚类分析中占比最大的一类作为代表构象。残基能量分解及聚类分析代表构象如图4所示。
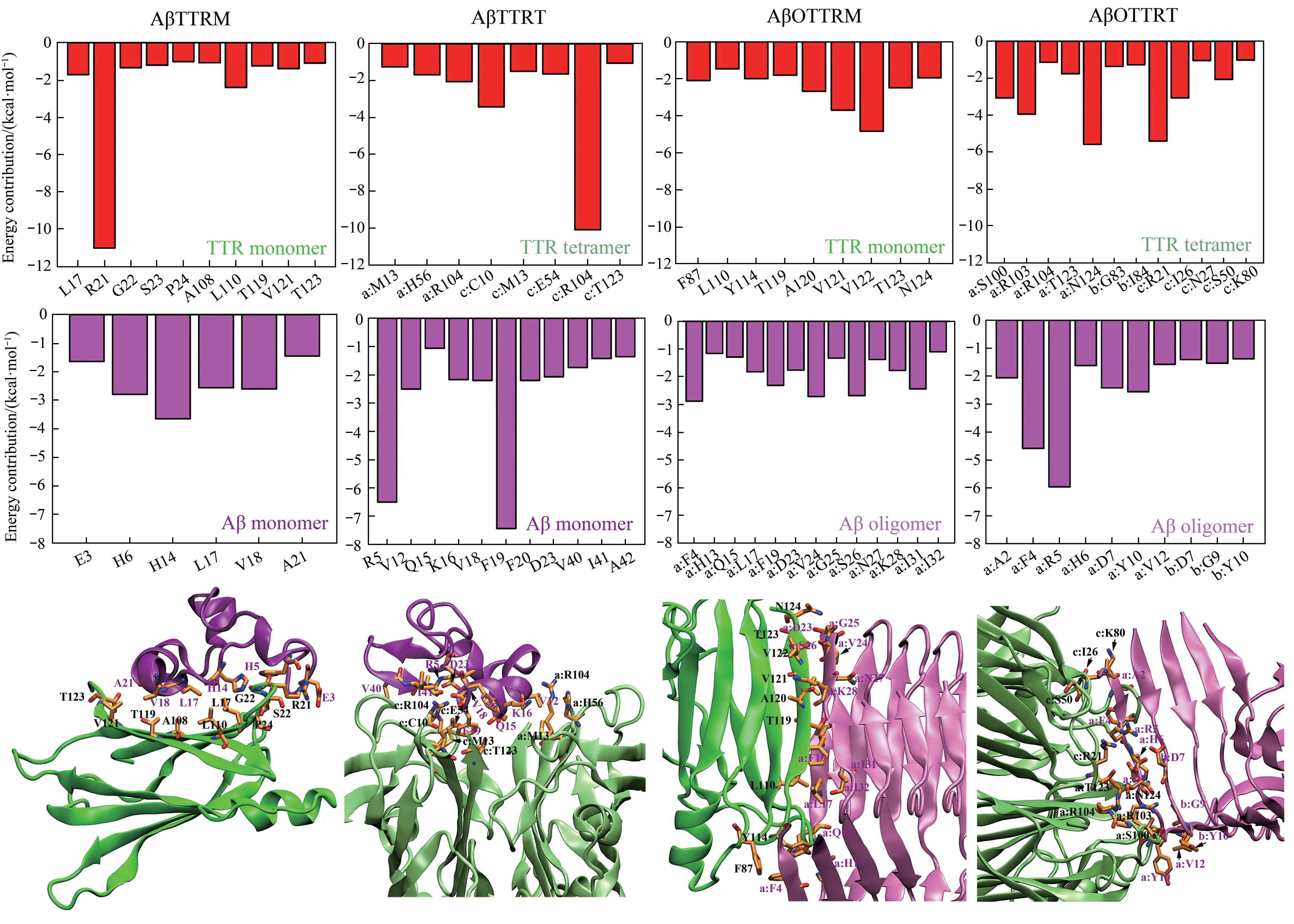
Fig. 4 The decomposition of free energy to residues and the representative conformations for system with the highest binding free energy
从图4中可以看出,AβTTRM体系结合自由能最高的模式为Aβ单体结合在TTR单体的DAGH片层界面,且TTR单体中与Aβ单体作用的残基主要为疏水残基,包括L17、P24、A108、L110、V121,在Aβ 单体6 个关键氨基酸中有3 个为疏水残基,分别为L17、V18、A21。这表明在该模式中疏水作用力是维持TTR单体与Aβ单体作用重要因素。值得注意的是,此前Du 等[24]通过测定TTR单体与Aβ单体复合物交联肽段的MS/MS谱确定TTR 单体的β 折叠链A 及周围残基是Aβ 单体的作用位点之一,而Aβ 单体中的中心疏水区(Aβ17-24)是与TTR单体作用的主要区域。Du等[24]报道的TTR 单体作用位点与图4 中Aβ 单体在TTR单体中的作用部位接近(DAGH 片层包含β 折叠链A),且该模式Aβ 单体参与作用的关键残基(L17、V18、A21) 属于中心疏水区残基。在AβTTRT体系中,模拟过程中结合自由能最高的模式为Aβ单体结合在T4结合位点的通道中,通过残基结合自由能分解得到的关键残基包括M13、E54、H56、R104、T123 等。T4 结合位点作为Aβ单体的结合位点也曾被Li 等[45]报道。他们通过NMR实验方法监测TTR四聚体与Aβ单体作用过程中酰胺-质子的化学位移,发现二者在作用过程中能够导致残基M13、V16、A109、L110、A120、V121以及V122发生明显的化学位移变化,这些残基均属于T4 结合位点或附近残基。尽管本工作通过残基自由能分解识别的关键作用残基与Li 等报道的残基有所差异,但这些残基基本位于T4 结合位点附近,其中M13 为报道的T4 结合位点残基。推测本工作模拟得到的TTR四聚体中与Aβ单体作用的关键残基与Li 实验所得残基之间的差异可能是由Aβ单体的构象差异引起的,因为Aβ作为无固定结构多肽,具有结构多样性的特点。但本工作识别的Aβ 结合位点与Li 等[45]通过实验确定的位点基本一致,即T4结合位点通道可作为“扣押”Aβ单体的位点。
相较而言,Aβ 低聚体与TTR 单体和四聚体的作用与Aβ 单体体系有较大的差距。从结构形态上来说,与Aβ单体的无序结构不同,Aβ低聚体主要为富含β折叠片层的有序结构。从结合自由能计算来看,AβOTTRM体系的3个结合模式仅有Model1中的TTR单体与Aβ五聚体维持了相互作用的高亲和力。通过对这一模式的结合自由能进行残基能量分解得到TTR 单体中的关键残基为T119、A120、V121、V122、T123、N124(图4),这些残基主要位于TTR单体的β折叠链H,且β折叠链H与Aβ五聚体的β 折叠边链(包括残基D23、V24、G25、S26、N27和K28)形成了稳定的β片层结构。一方面,根据Aβ 聚集过程中的Dock-Lock 机理[46],TTR 单体与Aβ 五聚体之间β 折叠片层结构的形成能够阻止游离Aβ 单体的进一步聚集。值得注意的是,近期Sun 等[47]通过分子动力学模拟的方法揭示αB晶体蛋白可通过覆盖Aβ聚集体及纤维结构中暴露的β折叠延伸面(即β折叠边链)抑制Aβ的聚集,这与本工作的研究结果一致。除此之外,Aβ聚集体中与TTR 单体作用的关键残基基本位于Aβ的13~28 残基区间,这与Gimeno 等[48]报道的Aβ12-28片段是Aβ 识别TTR 的关键区间的结论基本一致。TTR单体中除上述β折叠链H上的关键残基外,F87、L110以及Y114在与Aβ五聚体作用过程中也有较突出的贡献,表明这些残基同样能够影响TTR 单体与Aβ 聚集体之间的相互作用;并且Du等[28]也曾报道相比野生型TTR,Aβ与F87M/L110M突变TTR 单体之间具有更高的结合能力,表明残基F87 及L110 能显著影响TTR 与Aβ 之间的相互作用。
最后,通过分析Aβ 五聚体与TTR四聚体之间的相互作用发现原本打分结合能最高的Model1在模拟过程中结合自由能降低至-16.96 kcal/mol,而Model2的打分自由能由原来的-10.45 kcal/mol 增加至-31.18 kcal/mol。此外,从图2b的RMSD曲线及图4 AβOTTRT体系的代表结构发现,Model2中Aβ 五聚体的结构发生了较大的变化,五聚体N 端的β 折叠片层与其下层β 片层有明显错位,即在该模式中Aβ五聚体通过构象调整获得了与TTR四聚体较强的结合自由能。但值得注意的是,尽管Aβ五聚体构象N段上下片层之间的错位,聚集体的β折叠结构较好地被保留,表明该模式下TTR 四聚体并不破坏Aβ低聚体的二级结构。
3 讨论
自AD被报道以来,AD的发病机制及抗AD药物设计一直备受学界关注,然而截至目前仍没有有效的预防及治疗策略。TTR对AD所表现出的神经保护作用为AD的治疗提供了一种靶向蛋白质-蛋白质相互作用的药物设计策略[21,23]。本工作聚焦TTR对AD的神经保护作用,针对性地探究了不同结构形态的TTR和Aβ之间的相互作用。
本工作通过蛋白质-蛋白质对接的方式获得了Aβ 单体及聚集体分别与TTR 单体和四聚体可能的作用模式,并进一步通过分子动力学模拟探究了TTR 与Aβ 相互作用的动态过程。研究结果表明,TTR 的单体和四聚体均能与Aβ 单体产生较强的相互作用。针对TTR单体与不同构象的Aβ单体之间的相互作用,分子对接获得的自由能打分整体较高,且能在分子动力学模拟过程中维持较高结合亲和性(表1)。在本工作重点分析的3 个TTR 单体与Aβ 单体的作用模式中,Model2和Model3的结合位点曾被报道(图2)。在结合模式Model2中,Aβ 单体结合在TTR 单体的DAGH β 折叠片层,该折叠片层是组成TTR 四聚体的二聚体-二聚体作用界面,即该部位被包裹在TTR四聚体内部。并且,在分子动力学模拟过程中Model2在3 个模式中的结合自由能最高,为-41.48 kcal/mol。这一模式下,TTR 单体与Aβ 单体动态作用过程中的关键残基包括L17、 R21、 G22、 S23、 P24、 A108、L110、T119、V121、T123,这些残基中L17、R21位于β 折叠链A 上,G22、S23 属于AB loop 区残基,A108、L110 位于β 折叠链G 上,V121 位于β折叠链H上,且TTR的β折叠链A和β折叠链G也曾被报道是与Aβ作用的重要位点[24,28]。AβTTRM体系中,Model3结合模式中Aβ 单体主要作用于TTR 单体的EF 螺旋区及其周围残基,这一位点被报道为另一个Aβ 的作用位点[24,49]。他们推测,EF 螺旋及EF loop 可能是Aβ 的感应部位,能够探测并结合体内的Aβ,而Aβ作用于该部位能够进一步诱导TTR 四聚体的构象变化,并导致四聚体内部Aβ 作用位点(内部β 折叠片层,即DAGH 折叠片层)的暴露,从而结合更多的Aβ[49]。
在Aβ单体与TTR四聚体的研究体系中,同样有两个结合模式与实验报道较为一致,分别为Model1和Model3(图2)。结合模式Model1中,Aβ单体结合在T4结合通道中,该结合模式在模拟过程中维持了较高的亲和力,为-43.45 kcal/mol(表1)。TTR四聚体的T4结合位点及其周围残基作为Aβ单体作用部位曾被Li等[45]报道。在Model3中,Aβ单体则结合在TTR四聚体T4结合通道侧面的凹槽部位,该凹槽由四聚体中上下两个TTR 单体的EF螺旋结构包围组成,这在一定程度与EF螺旋及EF loop可能是Aβ感应部位的推测相符合[49]。此外,该部位也是视黄醛结合蛋白在TTR 四聚体结构中的结合部位[42],且视黄醛结合蛋白被报道能够参与TTR抑制Aβ聚集的过程[43]。
相较于Aβ单体与TTR的相互作用,Aβ聚集体与TTR 的相互作用有显著的差异。从结构上来说Aβ 单体为无定形非结构多肽,具有结构柔性大、体积相对小的特点,有利于在相互作用过程中通过构象的适当调整获得相对稳定的结合状态。相比之下,Aβ低聚体主要以富含β折叠结构的形式存在,结构固定且聚集体体积大,在作用过程中不易进行构象调整。本工作通过蛋白质-蛋白质对接获得的Aβ 五聚体与TTR 单体及四聚体的作用模式中,亲和力较高的模式较少。在AβOTTRM 体系中,仅Model1有较高的亲和力,且在模拟过程中能够持续稳定地作用(-45.47 kcal/mol)。通过分析Model1的特点可以发现该模式下相互作用主要存在于TTR 单体的β 折叠链H 与Aβ 五聚体的β 折叠边链,并且二者在模拟过程中形成了稳定的β折叠片层结构(图4)。Model2和Model3的对接打分及动力学模拟过程中的结合亲和性均较低,结合较弱。在Aβ五聚体与TTR四聚体作用体系中(AβOTTRT),尽管Model1在对接打分中有较高的亲和力打分(-36.28 kcal/mol),但该模式在模拟后的结合自由能仅为-16.96 kcal/mol,此外,虽然Model2的亲和力在动态相互作用过程中有所增加(由对接打分的-10.45 kcal/mol增加至-31.18 kcal/mol),但整体的结合自由能相较于其他3个体系仍相对较低。基于此,推测TTR四聚体与Aβ聚集体之间的相互作用可能不是TTR神经保护作用的主要原因。
综合以上讨论,本文对Aβ与TTR之间的相互作用机制进行了推测:TTR 对Aβ 的抑制聚集作用展现在两个方面(图5)。
一方面,TTR四聚体和TTR单体都能与Aβ单体发生相互作用,这种相互作用能够起到“扣押”Aβ 单体的作用[45,50]。尽管Li 等[45]认为,正常情况下体内的TTR 四聚体含量远高于TTR 单体,因而TTR四聚体是“扣押”Aβ单体的主要结构形式。但在此前Du 等[24]的工作中,他们通过酶联免疫分析和交联实验得到结论,相比TTR 四聚体,Aβ单体能够结合更多的TTR单体。与Du等的结论相似,Jain 等[51]认为相比TTR 四聚体,其单体形式能更好地抑制淀粉样纤维的形成。结合先前实验报道,本文认为TTR 四聚体和TTR 的单体均有“扣押”Aβ单体的能力。在不同情况下,TTR“扣押”Aβ 单体的形式不同。正常情况下,TTR 以稳定四聚体形式存在,其浓度远高于TTR 单体,因而是“扣押”Aβ单体的主要形式,T4结合通道是Aβ单体结合的主要位点。此外,本工作及其他工作报道表明,TTR四聚体中除T4结合通道可作为Aβ的结合位点外,四聚体结构中的EF螺旋、EF loop区也能够与Aβ 作用。当Aβ 作用于EF 螺旋形成的结合部位后,能够诱导TTR 四聚体解离[49,52],导致其内部疏水结合位点的暴露(即TTR 单体的DAGH折叠片层)。由于TTR 单体更易与Aβ 单体结合,因而TTR 单体与Aβ 单体的作用能够进一步促进TTR四聚体的解离,这时TTR单体即成为“扣押”Aβ单体的主要形式。
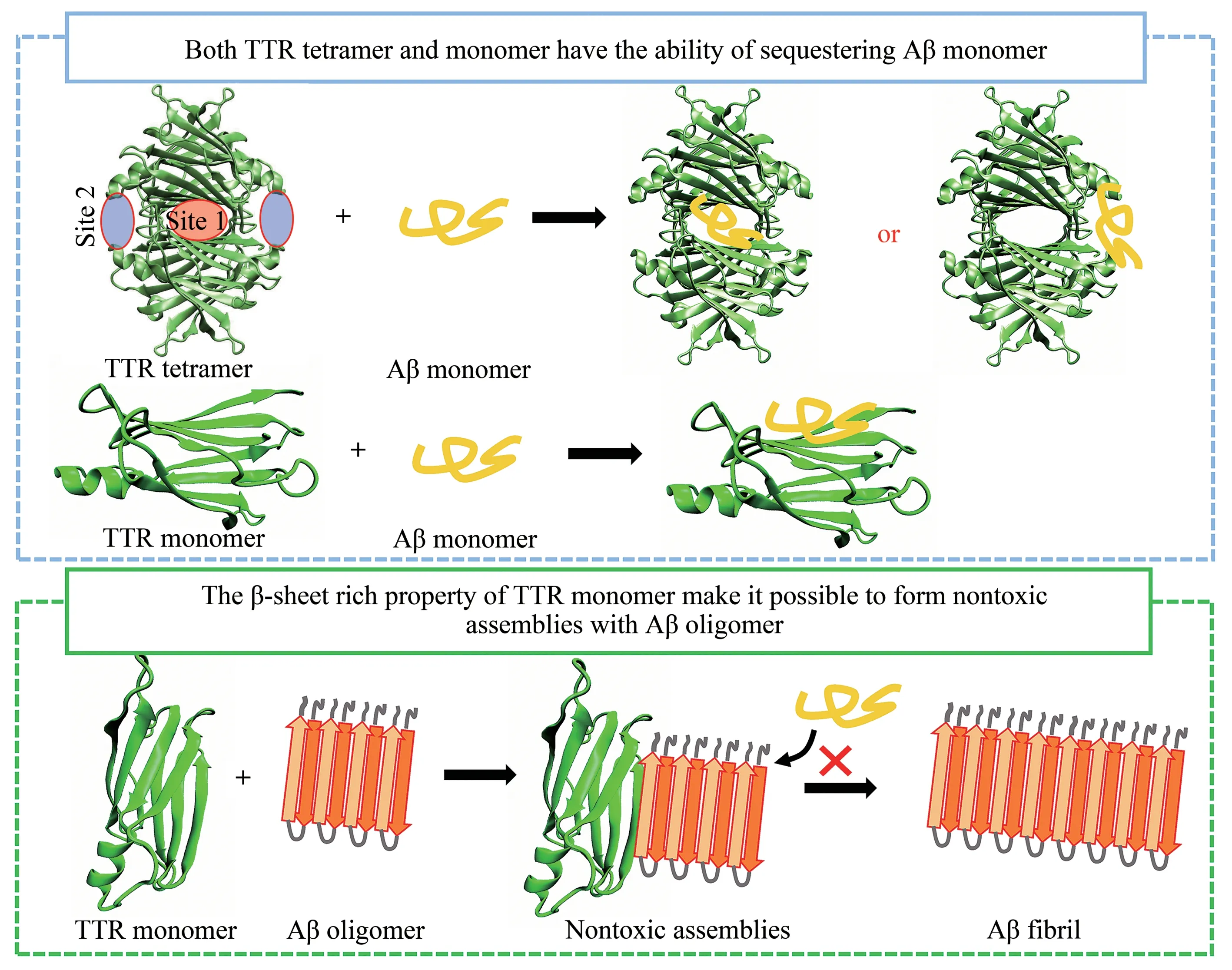
Fig. 5 The illustration of speculative interaction mechanisms between TTR and Aβ
另一方面,TTR 单体除了通过Aβ 单体的结合“扣押”其单体形式抑制Aβ聚集外,TTR单体特殊的结构特征使其能够与Aβ 的聚集体共聚集形成聚合度更高的聚集体。从结构上来说,TTR单体由8条β 折叠链组成了上下两层的β 折叠片层结构,分别为DAGH片层和CBEF片层,其中DAGH片层被包裹在四聚体界面围成了T4 结合通道。TTR 单体富含β 折叠片层结构的特征与Aβ 聚集体的结构相似,使得TTR 单体边缘的β 折叠链能够与Aβ 聚集体边缘的β 折叠链形成新的β 折叠片层结构而形成聚合度更高的无毒聚集体。重要的是,Garai等[25]通过双色荧光爆发重合检测实验确定TTR 单体能与可溶性Aβ 低聚体共聚集形成非纤维聚集体,且Cascella 等[53]通过原子力显微镜成像及细胞存活率检测(MTT)等实验方法发现TTR单体与Aβ聚集体作用形成的大聚集组装体的细胞毒性降低。这些实验结果无疑为上述推论提供了强有力的实验支撑。从这一机制推测出发,TTR单体是直接作用于Aβ 的毒性聚集体的结构形式,因此在一定程度上解释了Cao 等[19]报道的只有解离成单体的TTR 才能抑制Aβ 的细胞毒性。此外,根据淀粉样聚集的Dock-Lock 机理[46],当Aβ 聚集体的边缘β 折叠链被TTR单体占据后,聚集体募集游离Aβ单体的能力降低,因而能够阻止Aβ 的进一步聚集。本工作揭示的TTR 单体通过与Aβ 聚集体β 折叠边链作用从而抑制Aβ 聚集的机制与Sun 等[47]提出的αB 晶体蛋白通过覆盖Aβ 聚集体及纤维结构中暴露的β折叠延伸面(β 折叠边链)来抑制Aβ 聚集的结论一致。
基于研究结论,本文在此提出一些有关AD药物研发的思考:首先,TTR作为内源性蛋白在体内具有较好的生物相容性和低免疫性,因而以TTR为对象设计抗AD 策略具有得天独厚的优势。其次,本研究表明TTR 单体既能通过“扣押”Aβ 单体抑制Aβ 聚集,也能与Aβ 聚集体共聚集干扰Aβ的进一步聚集。因此,TTR单体可作为重点对象来开展抗AD药物的研究。一方面可以考虑增强TTR单体自身结构的稳定性,如设计稳定性氨基酸突变(T119M);另一方面可以考虑从TTR 单体结构出发增强TTR 单体对Aβ 单体及Aβ 聚集体的特异性识别作用,强化TTR单体与Aβ之间的相互作用。
4 结论
本工作聚焦于TTR对AD的神经保护作用,通过分子动力学模拟方法从分子层面探究了TTR 与Aβ 之间作用机制。综合本工作的研究发现得到以下结论。首先,TTR 结构的特殊性使得其对Aβ 单体具有较强的相互作用能力,可通过“扣押”单体的形式抑制Aβ 的聚集。这种结构特殊性表现为:a. 正常情况下TTR 以稳定四聚体形式存在,其结构中的T4结合通道及视黄醇结合部位为Aβ提供了适当的结合空间,有利于Aβ 单体的结合;b. 四聚体解离产生TTR 单体,导致疏水界面DAGH 折叠片层的暴露,使得Aβ 单体同样能够作用于该疏水界面(Aβ 单体的疏水核心为17LVFFA21),达到“扣押”Aβ单体的作用。其次,TTR单体的结构特征与Aβ 聚集体的结构相似,均为富含β 折叠的结构,这使得TTR单体能够与Aβ聚集体共聚形成聚合度更高的无毒聚集体,从而达到抑制Aβ 细胞毒性的效果。整体而言,本工作在原子尺度上探究了TTR与Aβ相互作用的机制,并揭示了TTR神经保护作用的分子机制,在一定程度上为基于TTR 神经保护作用机制的抗AD 药物研发提供了理论线索。
猜你喜欢
合成化学(2023年12期)2024-01-02
化工进展(2023年9期)2023-10-14
河南工业大学学报(自然科学版)(2021年6期)2022-01-26
化工科技(2021年5期)2021-11-24
温州大学学报(自然科学版)(2016年1期)2016-10-27
中国卫生标准管理(2015年1期)2016-01-14
河北科技大学学报(2015年6期)2015-03-11
河北科技大学学报(2015年5期)2015-03-11
应用化工(2014年7期)2014-08-09
石油化工(2014年1期)2014-06-07
