
氯沙坦钾原料药降解途径与降解杂质分析
2024-03-22 03:07潘静夏佳璇金一宝汤佳王冰黄晓龙
药学研究 2024年2期
潘静,夏佳璇,金一宝,汤佳,王冰,黄晓龙
(1.国家药品监督管理局药品审评检查大湾区分中心,广东 深圳 518017;2.深圳市药品检验研究院,国家药品监督管理局仿制药评价生物等效性研究重点实验室,深圳市药品质量标准研究重点实验室,广东 深圳 518057;3.中山大学药学院<深圳>,广东 深圳 518107)
氯沙坦钾(losartan potassium)作为首个临床非肽类血管紧张素Ⅱ受体阻滞剂,属于沙坦类药物,具有安全性高的特点,可同其他抗高血压药物一起使用,本品耐受性良好,不良反应轻微且短暂[1],临床上被广泛应用。2021年统计数据显示中国地区含氯沙坦钾活性成分药物制剂的销售额已达6.219 5亿元[2]。
原料药作为药品的主要活性成分,药物的有效性和安全性与原料药密切相关,药物因原料药生产、制剂过程中原料药与辅料发生化学反应及储存不当等因素均可能产生杂质从而降低药品活性,影响药物稳定性;甚至因各种原因产生的基因毒性杂质,会对人体健康产生严重的副作用和不良反应。如缬沙坦类产品中发现的N-二甲基亚硝胺及其他亚硝胺类基因毒性杂质[3-4],其来源于活性药物成分合成中产生,但也有研究表明沙坦类药物杂质来源于生产、贮存和运输等外部因素[5-6]。有研究将氯沙坦钾置于紫外、氧化及光照下对降解样本分析,发现样本具有慢性或急性毒性[7]。强制降解试验即在较短时间内采用剧烈条件使药物产生一定水平的降解[8],从而快速获取降解途径和降解产物,为药物安全性研究提供支持,以及对有关物质分析方法的开发,药物处方、包装材料选择和储存条件提供支持[9]。
本文依据人用药品技术要求国际协调理事会(ICH)指导原则等[10-12]开展氯沙坦钾原料药在氧化、强酸、强碱、高温、光照条件下的强制降解试验,开发基于高效液相色谱仪的分析方法,为氯沙坦钾原料药的降解途径和有关物质的研究奠定基础,为原料药的储存、制剂工艺、药品生产、运输和贮存条件提供参考。
1 仪器与试药
1.1 仪器LC-20A高效液相色谱仪(日本岛津公司);X500R UHPLC-Q TOF液质连用仪(美国Sciex公司);XS 105 DU天平(瑞士Mettler Toledo公司);BT 25S天平(德国Sartorius公司);HS3120超声仪(天津恒奥科技发展有限公司);SW23水浴锅(德国Julabo公司);KBF240恒温恒湿箱(德国Binder公司);UN110恒温干燥箱(德国Memmert公司)。
1.2 试剂试药氯沙坦钾原料药(浙江华海药业股份有限公司,批号:C5663-22-011);氯沙坦钾对照品(中国食品药品检定研究院,批号:100597-201703);《欧洲药典》氯沙坦杂质L(批号:LSTJ-12052019)、《欧洲药典》氯沙坦杂质M(批号:LSTJ-05262020)、氯沙坦杂质1(N-[[2′-(2H-tetrazol-5-yl)[1,1′-biphenyl]-4-yl]methyl]pentanamide,批号:LSTJ-05182020)均购于深圳市宏盛生物技术有限公司。乙腈(北京百灵威科技有限公司,色谱级);氢氧化钠(上海阿拉丁生化科技股份有限公司,分析纯);磷酸、盐酸和30%过氧化氢(广东广试试剂科技有限公司,分析纯);超纯水由Millipore超纯水仪自制。
2 方法与结果
2.1 液相色谱条件Waters XBridge BEH C18(4.6 mm×250 mm,5 μm)色谱柱,柱温30 ℃,流动相A为0.05%磷酸水溶液,流动相B为乙腈,流速1.0 mL·min-1,按照表1程序梯度洗脱,进样量10 μL,检测波长210 nm。

表1 液相梯度洗脱程序
2.2 液质检测条件Waters ACQUITY UPLC BEH C18(2.1 mm×100 mm,1.7 μm)色谱柱,柱温35 ℃,流动相A为5 mol·L-1乙酸铵(pH=4),流动相B为乙腈,流速0.5 mL·min-1,按照表2程序梯度洗脱。质谱信号采集时间0~20 min,电喷雾离子源,正离子模式,信息依赖采集(IDA)扫描模式。离子喷雾电压5 500 V,离子源温度500 ℃,去簇电压(DP) 80 V,雾化气和辅助气均为55 psi氮气,35 psi氮气作气帘气。扫描范围m/z50~1 000 Da,碰撞能(CE) 10 V。MS/MS扫描范围m/z50~1 000 Da,碰撞能(CE) 30 V。

表2 液质联用梯度洗脱程序

表3 氯沙坦钾降解产物及降解途径
2.3 溶液的制备稀释剂:乙腈-水(2∶8)。
供试品溶液:精密称取氯沙坦钾原料药12.5 mg,置于25 mL容量瓶,加稀释剂溶解并稀释制得0.5 mg·mL-1溶液,过滤,取续滤液作为供试品溶液。
对照品溶液:临用前将氯沙坦钾对照品于烘箱105 ℃烘干2 h,精密称取5 mg氯沙坦钾对照品,加稀释剂于2.5 mL容量瓶定容,作为对照品储备液;用稀释剂按比例稀释使用。
2.4 强制降解试验
2.4.1 氧化破坏室温环境下,精密称取氯沙坦钾原料药10 mg,置于玻璃具塞试管中,分别加入500 μL不同浓度的过氧化氢溶液(0、0.1%、3.0%、10%、30%),于室温下放置24、48、72、168 h后,加稀释剂定容至20 mL,摇匀后过滤(0.22 μm尼龙滤膜),取续滤液检测。精密称取氯沙坦钾原料药10 mg,分别加入500 μL不同浓度H2O2溶液(0、0.1%、1.0%、3.0%、10%、20%、30%),置于60 ℃水浴[13]中反应1、3 h,冷却后加稀释剂同法操作检测。
2.4.2 酸破坏精密称取氯沙坦钾原料药10 mg,置于玻璃具塞试管中,加入500 μL不同浓度盐酸溶液(0、1、2 mol·L-1),分别于60 ℃反应3、6 h及室温反应8 h。分别加500 μL对应浓度的氢氧化钠溶液中和,加稀释剂同法操作检测。
2.4.3 碱破坏精密称取氯沙坦钾原料药10 mg,置于玻璃具塞试管中,分别加入500 μL不同浓度氢氧化钠溶液(0、2、4 mol·L-1),分别于60 ℃反应3、6 h及室温反应8 h。分别加500 μL对应浓度盐酸溶液中和,加稀释剂同法操作检测。
2.4.4 高温破坏固体状态下,取氯沙坦钾原料药平铺于玻璃培养皿中(厚度<1 mm),置烘箱105 ℃加热6、12、24 h后,充分混匀后精密称取10 mg,加稀释剂同法操作检测。湿度考察,取氯沙坦钾原料药10 mg,置恒温恒湿箱中(55 ℃,RH 92.5%),放置24、72、120 h,加稀释剂同法操作检测。另取氯沙坦钾原料药10 mg,置恒温恒湿箱中(92.5 ℃,RH 25%),放置0.25、1、2 h后,加稀释剂同法操作检测。溶液状态下,称取氯沙坦钾原料药10 mg于10 mL离心管中,加稀释剂1.0 mL,水浴90 ℃加热破坏3、12、24 h,加稀释剂同法操作检测。
2.4.5 光照破坏取氯沙坦钾原料药平铺于玻璃培养皿中(厚度<1 mm),置恒温恒湿箱(25 ℃,RH 25%),(4 500±500) Lx白光,照射1、10、30 d,混匀精密称取10 mg,加稀释剂同法操作检测。另称取氯沙坦钾原料药10 mg于离心管中,加稀释剂1.0 mL,在(4 500±500) Lx照射1、7、20 d,加稀释剂同法操作检测。
2.5 降解试验结果
2.5.1 氧化破坏氯沙坦钾原料药在氧化(室温和加热)环境下均会发生不同程度的降解,1% H2O2(加热)环境中已出现降解产物,但其峰面积小于0.1%,在高于3% H2O2氧化破坏下,室温和加热环境均会发生降解。室温环境,氯沙坦钾的降解率和降解产物个数随H2O2浓度增加而增加;同浓度条件下,降解率和产物数随放置时间增加而增加。当氧化破坏伴随着高温环境(60 ℃),短时间内降解率可超过10%,伴随H2O2浓度的增加及时间的延长,最高降解率可达53.05%,产生29个降解产物。
2.5.2 酸破坏氯沙坦钾在酸室温条件下,最高降解率为4.84%。在酸伴随加热环境下,降解率在16.49%~37.97%,说明氯沙坦钾在酸降解中受温度影响大,温度越高其降解越明显。氯沙坦钾在2 mol·L-1盐酸中降解率低于1 mol·L-1盐酸,推测可能因氯沙坦钾在酸性环境中溶解度下降[13]所致。同浓度酸条件下加热时长对氯沙坦钾的降解影响不大,但降解产物稍有差异,相对保留时间(RRT) 2.20和2.29这两个降解产物相对含量随降解条件(酸浓度和时长)增强而增加。
2.5.3 碱破坏氯沙坦钾在碱室温环境微弱降解,RRT 0.136处的降解产物的相对含量随着碱浓度的增加而降低,推测因氯沙坦钾在碱环境中溶解度差异所致。另2 mol·L-1氢氧化钠比4 mol·L-1氢氧化钠新产生了RRT为2.20和2.29两个降解产物,与酸破坏中显著增加的降解产物一致,但在碱破坏中仅占0.2%左右,远低于酸环境。同浓度碱环境下加热时长对降解率影响不大。初步判定氯沙坦钾对碱性环境相对稳定。
2.5.4 高温破坏《中国药典》2020年版(二部)和相关文献[11,15]表明,氯沙坦钾原料药易吸潮,颗粒烘干后应立即压片,故分别考察氯沙坦钾在固体和液体状态下的高温破坏。氯沙坦钾原料药在固体高温干燥(烘箱105 ℃)环境12 h和高温低湿(92.5 ℃,RH 25%)环境2 h内稳定性良好。但在固体高温高湿环境(55 ℃,RH 92.5%)3 d降解率可超30%,第5天降解率达43.97%,但基于HPLC的紫外检测,只有4个降解产物(相对含量最高的仅为0.39%),推测其降解产物不具有紫外吸收。氯沙坦钾在溶液状态于高温水浴90 ℃环境24 h后降解率为7.36%,RRT 2.20和RRT 2.29这2个降解产物含量均随时间的延长而增加。
2.5.5 光照破坏考察氯沙坦钾在固体和溶液状态下对白光[25 ℃,RH 25%,(4 500±500) Lx]的敏感性,氯沙坦钾原料药在固体状态下10 d降解率可超10%,光照30 d降解产物数有4个,其中RRT 2.38降解产物含量随时间的延长而增加。氯沙坦钾溶液状态对光照敏感,仅1 d降解率达5.54%,20 d降解率达47.26%,产生8个降解产物。其中RRT 1.46在固体和溶液状态下含量均会随时间的延长而增加。
2.6 氯沙坦钾降解途径以氯沙坦钾降解率和峰面积大于主峰0.1%的降解产物个数进行比较,将67种降解条件下的降解率和降解产物个数作气泡图(见图1)。对氯沙坦钾原料药在氧化、酸、碱、高温和光照条件下的降解率进行总结。氯沙坦钾在氧化(室温和加热)环境中均呈现极大的降解率,H2O2浓度、氧化环境温度以及氧化时长均有影响。在酸破坏环境中降解率受温度影响大,酸加热环境的降解率远高于其在室温环境的降解率。虽然碱环境相对稳定,但其降解同样受温度影响,碱加热环境中降解率有所增加。高温高湿环境下降解率也远高于高温干燥环境。一旦配制成为溶液状态,光照条件下降解率急剧上升。
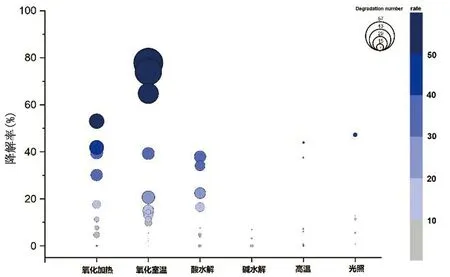
图1 氯沙坦钾原料药在67种降解条件下的降解率和降解产物气泡图
2.7 氯沙坦钾降解产物分析氯沙坦钾原料药在多种强制破坏条件下均产生了22种降解产物(峰面积>0.1%氯沙坦钾峰面积,见图2),结合液质联用对氯沙坦钾的母核结构和碎片分子量以及降解杂质的二级断裂碎片的测定结果,推测了降解产物中的19种化合物的分子量和分子式,确定了峰1、8、19和20这4个降解杂质的分子结构。
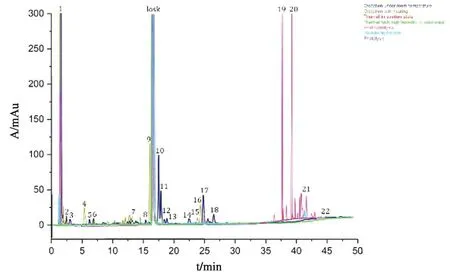
图2 氯沙坦钾各降解条件下的降解产物色谱图
在氧化破坏中,峰1峰面积为主峰(氯沙坦钾)峰面积的3倍左右(见表1),如图3所示,在正离子模式下,峰1的一级质谱(MS1)提示503.242 4、252.125 1、235.097 5、207.091 3,503.242 4可能为其准分子离子,对m/z503.242 4、252.125 1、235.097 5、207.091 3分别进行二级碎片采集,发现m/z252.125 1的碎片中,含有m/z235.097 5、207.091 3,结合m/z503.242 4可能为[2M+H]+,判定[M+H]+准分子离子峰为m/z252.125 1,二级质谱(MS2)提示[M+H]+脱去-NH3后,形成m/z235.098 0的[M+H-NH3]+,随后四氮唑环断裂离去N2,与苯成环结合成苯并吡唑形成m/z207.092 0,吡唑环进一步断裂形成m/z180.079 5碎片离子。

图3 RRT 0.140降解杂质质谱裂解
峰8同样作为氧化降解产物,如图4所示,在正离子模式下,其MS1显示[M+H]+准分子离子峰m/z336.180 9,MS2含有235.096 1、207.089 3,提示其母核与峰1均含有四氮唑联苯结构,且根据氯原子的质谱规律,峰1和峰8中均不含Cl原子。氯沙坦钾结构包括咪唑环、联苯以及四氮唑3部分,结合降解产物的碎片离子,认为在氧化条件下O=O最先进攻氯沙坦钾的咪唑环,Cl原子离去同时保留联苯和四氮唑部分,初步推断咪唑环在氧的进攻下断裂,发生1,4-环加成,离去-N=C-Cl形成化合物峰8分子式C19H21N5O(分子量335.17);峰1是峰8离去烷基支链后进一步的氧化降解产物,分子式C14H13N5(分子量251.12)。文献[16]表明,氯沙坦钾的咪唑环易发生单线态氧的光敏化反应,在氯沙坦钾的氧化混悬液中也发现了这两个成分。将其与对照品比对后证实峰1为(2′-(1H-tetrazol-5-yl)-[1,1′-biphenyl]-4-yl)methanamine,峰8为N-[[2′-(2H-tetrazol-5-yl)[1,1′-biphenyl]-4-yl]methyl]pentanamide,是缬沙坦去缬氨酸杂质。

图4 RRT 0.87降解杂质质谱裂解
峰19和20这两个降解产物在酸中的显著增加(峰面积占比达到了9.84%),在正离子模式下,峰19和峰20的MS1分别为827.319 3和827.321 5,MS2有809.305 4、405.155 1、377.150 5和207.090 5且丰度高,氯沙坦钾在酸性环境下其咪唑环会发生断裂[17],结合离子碎片初步判断两者为同分异构体,[M+H]+准分子离子峰为m/z827.32。通过对照品比对保留时间和二级碎片确认为《欧洲药典》(EP)收载的杂质L和杂质M(见图5~6)。

图5 RRT 2.21降解杂质质谱裂解

图6 RRT 2.29降解杂质质谱裂解
3 讨论
3.1 降解条件的选择基于ICH指导原则[10,18]设计了本实验的降解条件,如30%过氧化氢在超过60 ℃[12,19]的条件下分解速度会显著增加故选择了60 ℃作为氧化加热降解条件,联合氧化室温条件以考察缓慢氧化对降解的影响。文献[14]表明氯沙坦钾在酸性环境中会发生降解,故设置了高于常规考察的盐酸浓度2 mol·L-1和1 mol·L-1;未见有文献报道其在碱环境中的稳定性,故设置了高浓度的氢氧化钠4 mol·L-1和2 mol·L-1。高温降解试验根据应用,分别考察了其在固体状态下高温低湿、高温高湿、高温干燥以及溶液状态下高温水浴4种条件。
3.2 氯沙坦钾降解途径基于氧化、酸、碱、高温以及光照强制降解实验结果,氯沙坦钾对氧化、酸、高温和光照环境敏感。在氧化环境中降解率最高(77.94%),后依次是光照(47.26%)、高温(43.97%)和酸破坏(37.97%)。氧化环境中产生的降解产物最多为57个,其次是酸降解有23个。结合氯沙坦钾和降解杂质的碎片离子,初步得出以下的裂解规律,具有氯沙坦钾母核结构的杂质会产生m/z405、377、235、207、190碎片离子峰;咪唑环片断和联苯片断与四氮唑环片断容易断裂,产生碎片离子,MS有207和235时提示含联苯并四氮唑结构;咪唑环和四氮唑连接的基团容易断裂,可据此推断咪唑环和四氮唑上的取代基团。基于此裂解规律,我们对降解产物中的22种成分进行了结构解析,确定了峰1、8、19和20 4个化合物的结构,总结了峰1和峰8这两个氧化降解杂质的裂解规律。氯沙坦钾结构中咪唑环上的Cl原子易受到氧化离子的攻击,咪唑环断裂的同时,N原子帮助结构的重排,而产生氧化降解产物。此外,咪唑环对热和光照环境不稳定,氯沙坦钾的咪唑环上连接的Cl原子和-CH2OH可能是氯沙坦钾在溶液中对光和高热不稳定的原因之一。四氮唑是羧基的生物电子等排体,与羧基具有十分相似的理化性质,具有较强的酸性,但与联苯结构可形成较稳定的复合物,推测可能是其对碱相对稳定的原因。
氯沙坦钾的降解途径主要为氧化破坏、酸破坏和溶液高温破坏,同时因其易吸潮的特性,固体状态下高温伴随高湿环境、溶液状态下白光(25 ℃,RH 25%)照射也存在显著降解。因此,在氯沙坦钾原料药产品包装储存及药物制剂处方设定时应避免接触氧化剂及酸,其常规制剂工艺也宜采用粉末直压、干法制粒等,同时需控制环境温、湿度及避免强光。通过对氯沙坦钾降解途径的研究,有助于探索药品贮藏及药品制剂生产中的杂质变化,用于氯沙坦钾原料药及其制剂的质量控制。
4 结论
本试验建立了一种在各种降解条件下能有效分离氯沙坦钾和其降解杂质的HPLC分析方法,经方法学验证此分析方法稳定可靠。考察了氧化、酸、碱、高温、光照5大类强制降解条件对氯沙坦钾原料药的影响,共设计67种降解条件,明确了氯沙坦钾的降解途径主要为氧化破坏、酸破坏和固体高温高湿破坏,溶液高温以及溶液白光破坏。本研究可为氯沙坦钾原料药的质量控制尤其是有关物质的研究提供参考,也可为氯沙坦钾原料药的贮藏及制剂工艺研究提供参考依据。
猜你喜欢
健康体检与管理(2022年2期)2022-04-15
中国药学药品知识仓库(2022年5期)2022-04-11
云南化工(2021年7期)2021-12-21
核化学与放射化学(2020年4期)2020-08-21
中国盐业(2018年20期)2019-01-14
湿法冶金(2018年2期)2018-04-25
质谱学报(2016年4期)2016-08-02
中国卫生标准管理(2015年8期)2016-01-15
实用中西医结合临床(2015年7期)2015-02-28
中华皮肤科杂志(2014年4期)2014-12-19
