
TPI缺乏症斑马鱼模型的构建及分析
2024-03-22 06:54:58孙飘李颖刘帆王璐
遗传 2024年3期
孙飘,李颖,刘帆,王璐,2
研究报告
TPI缺乏症斑马鱼模型的构建及分析
孙飘1,李颖1,刘帆1,王璐1,2
1. 中国医学科学院血液病医院(中国医学科学院血液学研究所),实验血液学国家重点实验室,国家血液系统疾病临床医学研究中心,细胞生态海河实验室,天津 300020 2. 天津医学健康研究院,天津 301600
磷酸丙糖异构酶缺乏症(triosephosphate isomerase deficiency,TPI DF)是一种严重的多系统退行性疾病,通常表现为溶血性贫血、神经肌肉功能障碍和易感染,患者多于起病5年内死亡。目前尚不清楚TPI DF的具体发病机制,缺乏有效的临床治疗方法。本研究选取TPI DF患者中最常见的突变位点E105D,构建了表达人源性E105D(hE105D)的转基因斑马鱼()模型[Tg(:E105D-eGFP)]。功能分析表明,过表达E105D影响红系及髓系细胞发育、导致神经以及肌肉发育异常。综上所述,本研究构建了磷酸丙糖异构酶缺乏症的斑马鱼疾病模型,并能够复现TPI DF患者的大部分临床表型,该模型为后续研究TPI DF的发病机制及药物筛选提供了新的实验动物模型。
磷酸丙糖异构酶缺乏症;E105D;斑马鱼;疾病模型
磷酸丙糖异构酶(triosephosphate isomerase,TPI)是由位于染色体12p13的基因编码的糖酵解酶[1],在所有组织细胞中均有表达。两个TPI单亚基形成同型二聚体,发挥催化酶活性,负责磷酸二羟丙酮(DHAP)和3-磷酸-甘油醛(G3P)之间的相互转化[2]。糖酵解途径、磷酸戊糖途径、糖异生和脂代谢等过程可以通过DHAP或G3P相互连接[3]。因此,TPI可以通过调节DHAP和G3P之间的相互转化从而调节代谢走向[2,3]。
磷酸丙糖异构酶缺乏症(TPI deficiency,TPI DF)是一种罕见的常染色体隐性遗传病,于1965年首次发现[4]。TPI DF属于最严重的糖酵解酶病,常发病于幼儿期[5]。该病的典型临床特征为溶血性贫血、免疫功能下降,伴随神经系统损伤、进行性肌肉功能障碍等症状,大部分患者会在幼儿期死亡[6]。遗传学证据表明基因突变会导致TPI DF[7],目前约有19种突变位点被鉴定报道,主要包括错义突变、无义突变和移码突变等。其中,E105D是发病率最高的一种变异[8],研究表明E105D会抑制TPI二聚体形成,降低蛋白的稳定性,从而致病[9,10]。由于TPI DF的具体发病机制尚不清楚,目前临床上缺乏有效的治疗方法。
斑马鱼()是一种小型脊椎动物,具有独特的生物学优势:繁殖发育快、体型小、实验周期短,适合大规模药物筛选;体外受精且胚胎透明,操作、观察方便,适用于活体成像等。此外,斑马鱼的发育过程与哺乳动物相似,基因组和信号通路与人类高度保守。因此,斑马鱼已成为研究神经、心血管、血液、骨骼肌肉和代谢等相关疾病的重要模式动物。
构建稳定遗传的疾病模型,不仅可以研究相关基因的功能,而且有助于探索相关疾病的发病机制,以及用于筛选新的药物治疗靶点。本研究利用转座子系统将人源性E105D基因序列整合到斑马鱼基因组中,构建了能够稳定表达人源性E105D的转基因斑马鱼品系[Tg(:E105D-eGFP)],该转基因品系具有贫血、免疫功能低下、神经肌肉发育异常等类似于人类TPI DF的表型,这为后续的分子机制研究和药物筛选提供了动物模型。
1 材料与方法
1.1 材料
pGEM-h质粒(HG16150-G)购自北京义翘神州公司。promotereGFP质粒为中国科学院动物研究所刘峰研究员团队惠赠。实验所使用的斑马鱼为Tübingen品系,成年斑马鱼用新鲜丰年虾喂养,饲养在上海海圣斑马鱼养殖系统中,水温28℃,pH值为7~7.5,电导率约为550 μS/cm。
1.2 质粒构建
利用NEB官网中的NEBuilder Assembly Tool,设计h片段和promotereGFP载体的引物,分别记为F insert和R insert、F vector和R vector。以pGEM-h质粒为模板,用F insert+R insert引物进行PCR,扩增得到含有同源臂的h片段。设计两条包含hE105D点突变的反向互补配对引物,记为G315C F和G315C R。以pGEM-h质粒为模板,分别用F insert+G315C R和G315C F+R insert这两对引物进行第一轮PCR,将扩增得到的产物按照1∶1混合作为模板,利用F insert+R insert进行第二轮PCR,扩增得到含有同源臂的hE105D片段。同时以带有promotereGFP序列的载体为模板,用F vector+R vector引物进行PCR扩增得到带有同源臂的promotereGFP载体片段。最后利用DNA重组试剂盒(E2621L,美国New England Biolabs公司)将含有同源臂的h和hE105D片段分别与载体片段进行同源重组,得到:heGFP和:hE105DeGFP质粒。引物序列见附表1。
1.3 整胚原位杂交
整胚原位杂交步骤按照参考文献[11]进行。不同时期的斑马鱼胚胎,经4%多聚甲醛固定过夜,于–20℃甲醇脱水至少2 h后梯度复水。根据不同的发育时期,用蛋白酶K(39450-01-6,美国Sigma-Aldrich公司)进行适度通透。清洗与预杂交后加入探针,于分子杂交炉65℃杂交过夜,用SSCT(3 mol/L NaCl、0.34 mol/L C6H5Na3O7·2H2O、0.1% Tween-20),MABT (0.1 mol/L马来酸、0.15 mol/L NaCl、0.1% Tween-20)依次漂洗,后加入封闭液孵育1 h,加入抗地高辛抗体(11093274910,美国Roche公司),4℃过夜孵育。次日用MABT和BCL (0.1 mol/L Tris-HCl(pH 9.5)、0.1 mol/L NaCl、0.05 mol/L MgCl2、0.1% Tween-20)漂洗,最后加入BM Purple (11442074001,美国Roche公司)染色。特异性信号显示后终止染色,固定后加入甘油于4℃保存。原位杂交实验结果使用ImageJ软件分析[12]。本研究检测基因所需探针的引物序列见附表2。
1.4 蛋白免疫印迹
取约50枚受精后24小时(hours post fertilization,hpf)的斑马鱼胚胎,吸干水分后加入蛋白裂解液,研磨器研磨提取蛋白,离心后取上清,BCA法对蛋白进行定量。取40 μg变性后的蛋白在浓度为12% SDS-PAGE凝胶中电泳分离蛋白,然后湿转至PVDF膜上。5% BSA孵育1 h后,加入TPI1抗体(AF8214,1∶1000,上海碧云天生物技术有限公司),4℃孵育过夜。次日TBST清洗3次后,加入兔源二抗(111-035-003,1∶10000,美国Jackson公司)孵育2 h,TBST清洗3次后加入显影液,放入化学发光成像仪中进行显影。
1.5 鬼笔环肽染色
收集所需斑马鱼胚胎后,4%多聚甲醛固定过夜,次日用乙醇脱水2 h后进行梯度复水,蛋白酶K通透后固定,PBST清洗4遍后加入鬼笔环肽染液(A12379,1∶400,美国Thermo Fisher Scientific公司),室温孵育1 h后终止染色,使用共聚焦显微镜拍照。
1.6 邻联茴香胺染色
收取斑马鱼胚胎,加入由2.7 mmol/L邻联茴香胺溶液(BNN2362,美国Sigma-Aldrich公司)、0.01 mol/L醋酸钠溶液(pH4.5)和0.7%过氧化氢溶液混合而成的染液,室温避光染色20 min。染色充分后,将染液吸出,双蒸水清洗胚胎,用4%多聚甲醛固定后拍照。
1.7 吉姆萨染色
取20枚受精后2天(days post fertilization,dpf)斑马鱼胚胎,剪尾后收集外周血至PBS溶液中,并转移适量混合液至载玻片上,离心后使用甲醇固定2 min,随后加入吉姆萨染液(G1010,北京索莱宝生物科技有限公司),室温染色20 min。待染色结束,冲洗晾干后拍照。
1.8 流式分析
取Tg (:dsRed)斑马鱼胚胎放入培养皿中,加入0.5%胰酶消化至单细胞状态,终止反应后离心并使用PBS重悬,经300目细胞滤网过滤得到单细胞悬液,用流式细胞仪(FACS canto II,美国BD Biosciences公司)进行分析。记录红细胞数目,根据使用的胚胎总数获得每个胚胎的红细胞数量。
1.9 TPI酶活性测定
TPI酶活性采用磷酸丙糖异构酶试剂盒(G0824W,苏州格锐思生物科技有限公司)进行测定。取斑马鱼胚胎,加入TPI提取液进行冰浴匀浆,离心后取上清液10 μL加入96孔板中,依据说明书加入相应试剂,随后使用酶标仪检测450 nm处光吸收增加量,即可得到TPI酶活性大小。
1.10 统计方法
应用GraphPad Prism8 软件进行数据分析。统计显著性分析根据数据类型选择检验方法:计数资料以百分率(%)表示,两组之间比较采用卡方检验;计量资料数据结果以平均值±标准差表示,比较采用检验。<0.05时认为差异显著。星号用来表示差异的显著性,具体为:*<0.05,**<0.01,***<0.001,****<0.0001,n.s.表示无显著性差异。
2 结果与分析
2.1 构建过表达人源TPI1和TPI1E105D转基因品系
2.1.1TPI1是致病性突变中最常见的变异
目前,已经报道了约19种导致TPI DF的基因突变类型(图1A),其中一些突变会导致TPI功能域受损进而影响其功能(表1)。TPI DF存在基因型-表型相关性,特别是作用于TPI蛋白二聚化位点的突变会导致更加严重的神经系统疾病[13]。其中,E105D突变位置靠近二聚化位点(图1B),是最常见的致病突变,占比约为80%[14]。目前研究报道了21例患者存在E105D突变,包括12例纯合突变和9例复合杂合突变。本研究总结了E105D突变患者的临床症状(附表3),发现E105D纯合突变的患者均存在严重的TPI DF症状,包括溶血性贫血、进行性的神经肌肉功能障碍、反复感染以及呼吸衰竭等;而E105D复合杂合突变的患者则会因另一突变类型的不同而存在表型差异:例如E105D/L29del2nt复合杂合突变患者存在严重溶血性贫血、反复肺部感染,而E105D/Q181P复合杂合突变患者则未发现溶血性贫血,主要存在神经肌肉相关的症状等。基于E105D是临床上最常见的突变位点,以及E105D纯和突变的患者均具有严重的TPI DF症状,所以本研究选择E105D构建疾病模型。
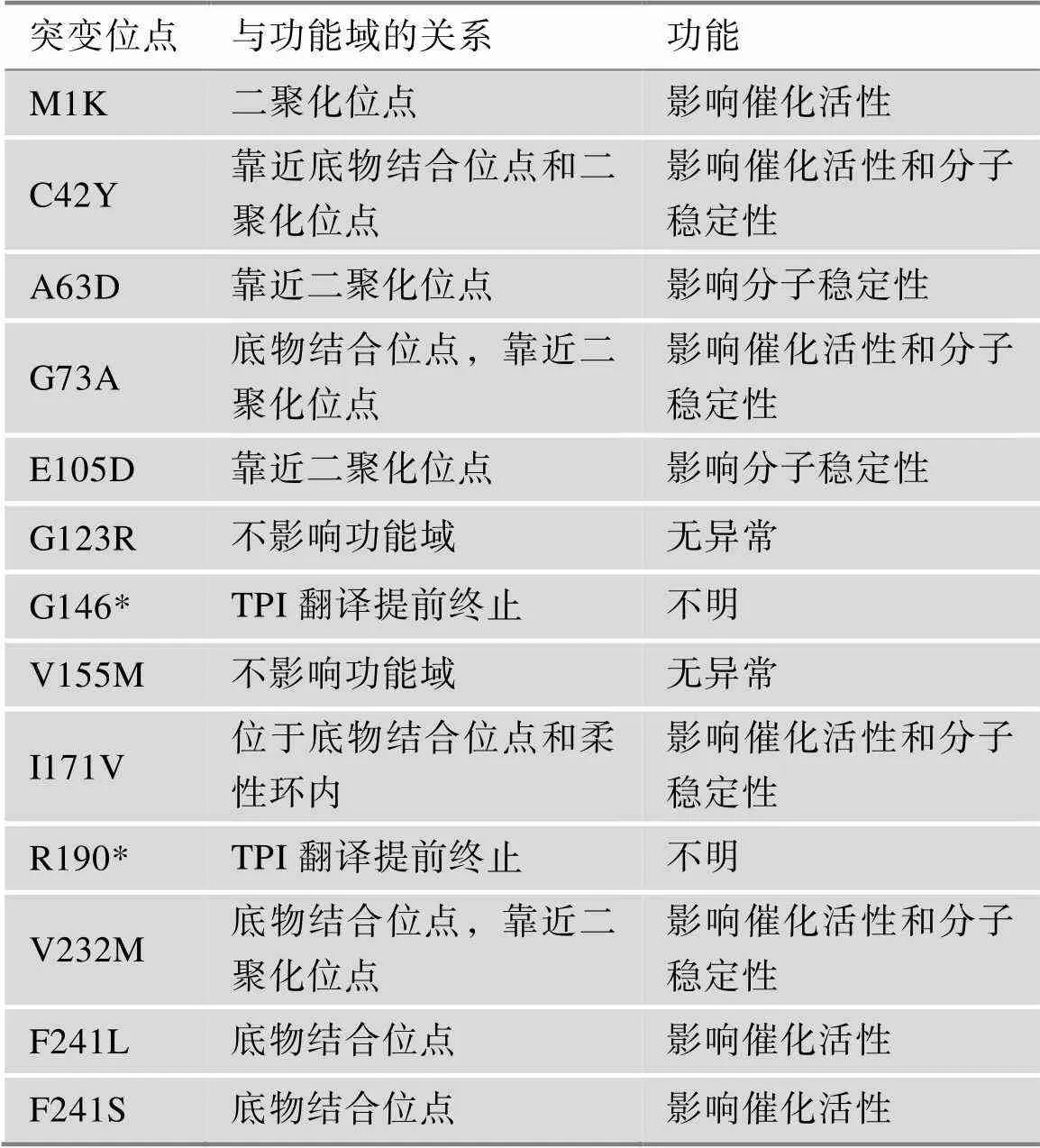
表1 TPI DF突变位点信息
2.1.2:h-eGFP和:hE105D- eGFP质粒构建
基于pGEM-h质粒,本研究首先通过PCR扩增获得h片段,同时应用定点突变方法得到hE105D片段,利用同源重组将两个片段分别连接到promotereGFP质粒中,获得: h-eGFP质粒以及:hE105D-eGFP质粒(图1C)。Sanger测序结果显示,与: h-eGFP质粒相比,:hE105D-eGFP质粒中目标位置碱基位点被成功突变(c.315G>C) (图 1D)。
2.1.3 Tg(:h-eGFP)和Tg(:hE105D- eGFP)品系的建立及验证
将上述构建的:h-eGFP质粒和:hE105D-eGFP质粒分别与体外合成的mRNA混合后注射至1-细胞期的斑马鱼胚胎中。在荧光显微镜下挑选带有绿色荧光的F0斑马鱼胚胎培养至性成熟。将F0嵌合体斑马鱼与野生型斑马鱼杂交,所得胚胎中选取有绿色荧光的F1代斑马鱼进行培养,得到稳定遗传的Tg(:h-eGFP)和Tg(:hE105D-eGFP)转基因斑马鱼后代(图2,A和B)。
为了进一步确定人源性基因h和hE105D的表达,本研究提取转基因斑马鱼子代胚胎蛋白进行免疫印迹实验。结果表明,与野生型相比,转基因胚胎中TPI1蛋白表达量明显增加(图2C),说明Tg(:h-eGFP)和Tg(:hE105D-eGFP)转基因斑马鱼胚胎可以实现TPI1蛋白过表达。TPI酶活性测定结果表明,Tg(:h-eGFP) 转基因斑马鱼胚胎中TPI酶活性显著增强,而Tg(:hE105D-eGFP)转基因胚胎中TPI酶活性则显著降低,说明E105D过表达可能降低斑马鱼体内TPI酶活性(图2D)。
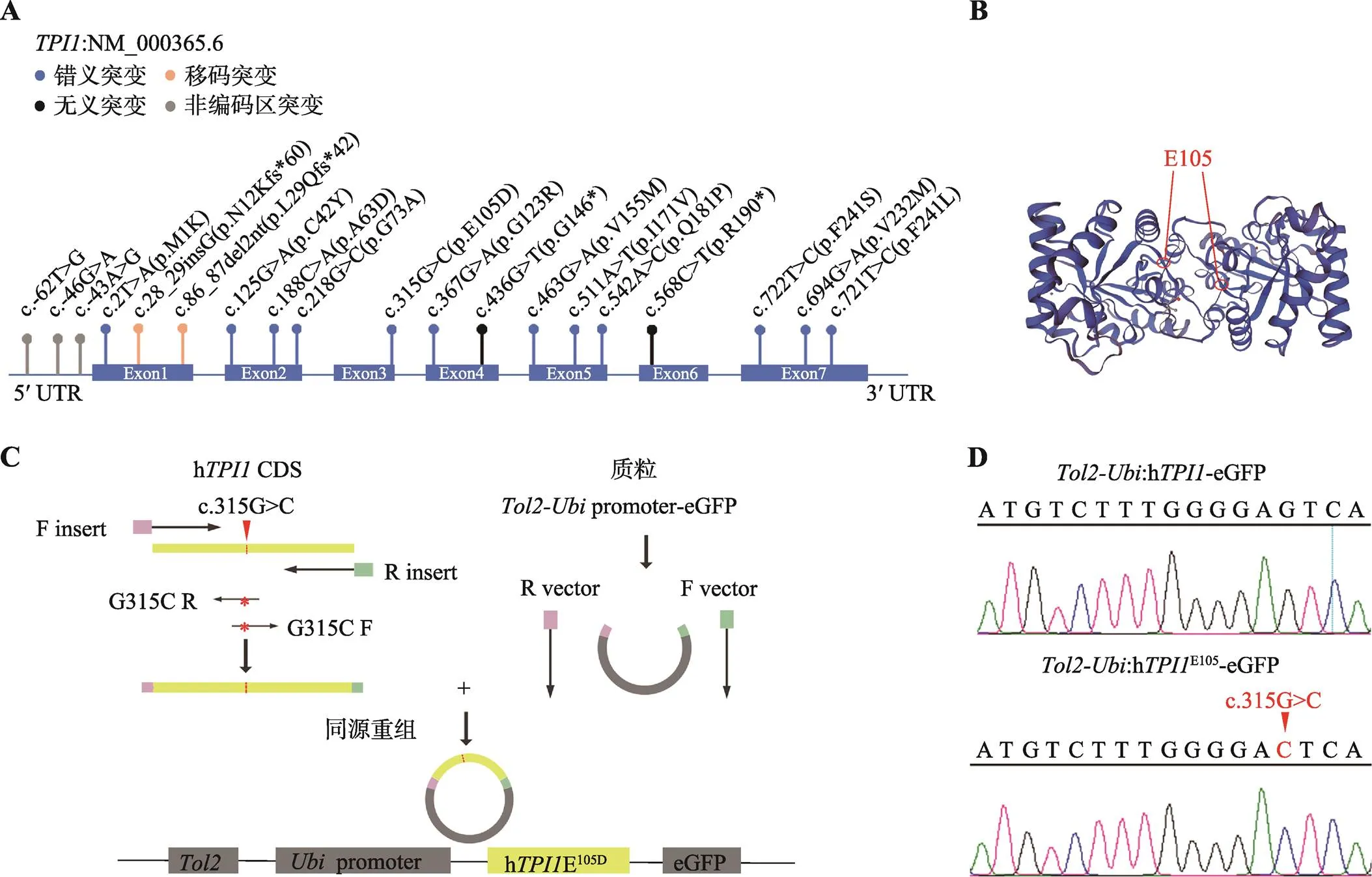
图1 Tol2-Ubi:hTPI1-eGFP和Tol2-Ubi:hTPI1E105D-eGFP质粒构建
A:基因示意图(图中标出了已报道的突变位点);B:TPI1 E105在TPI1二聚体中所处位置;C::hE105D-eGFP质粒构建流程;D::h-eGFP和:hE105D-eGFP质粒的测序鉴定结果。其中上图为对照组野生型基因序列,下图为带有E105D突变位点的序列,红色箭头代表突变位点。
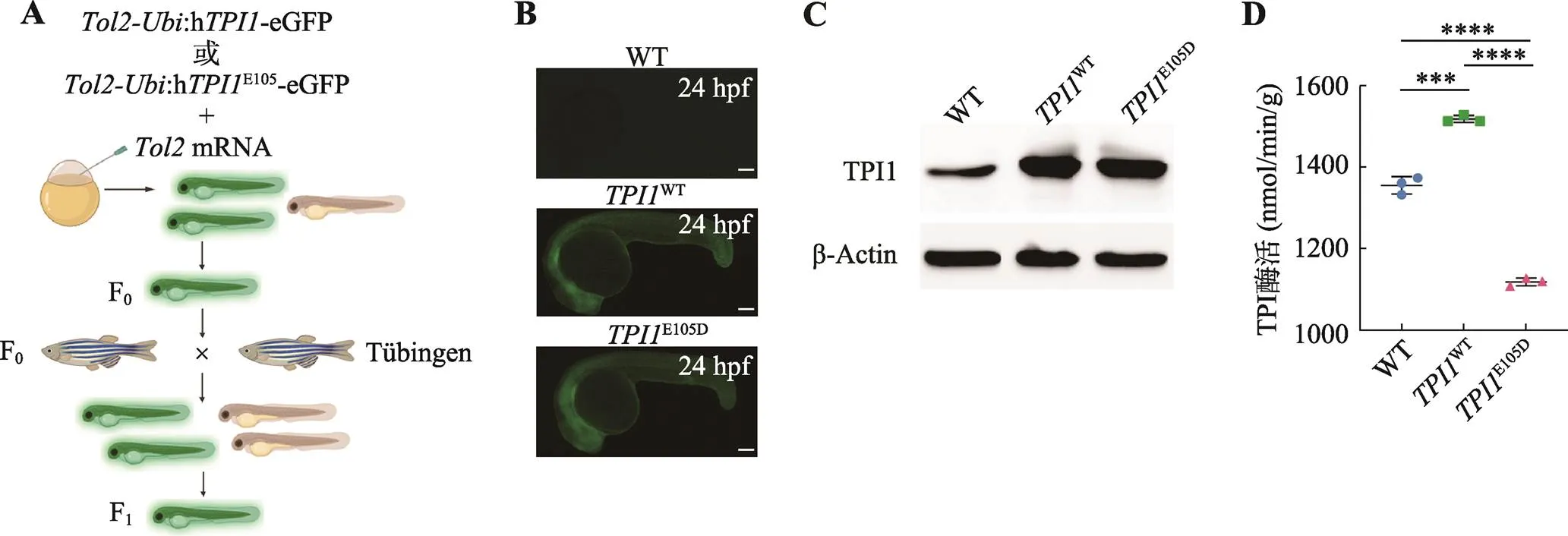
图2 Tg(Ubi:hTPI1-eGFP)和Tg(Ubi:hTPI1E105D-eGFP)转基因品系的构建
A:Tg(:h-eGFP)和Tg(:hE105D-eGFP)转基因品系的构建流程。F0代表注射质粒和转座子mRNA后有绿色荧光的嵌合体,F1代表能够稳定遗传的带有绿色荧光的子代;B:24hpf转基因斑马鱼胚胎荧光图片。比例尺为100 μm;C:野生型、Tg(:h-eGFP)和Tg(:hE105D-eGFP)转基因斑马鱼胚胎中TPI1蛋白表达情况;D:野生型组和转基因组TPI酶活。数据展示为平均值±标准差,*表示差异的显著性(***<0.001,****<0.0001)。值由双尾检验计算得出。WT为Tg(:h-eGFP)简写,E105D为Tg(:hE105D-eGFP)简写。
2.2 过表达人源TPI1E105D导致神经以及肌肉发育异常
TPI DF是一种进行性多系统退行性疾病,最终导致溶血性贫血、神经肌肉功能障碍、感染易感性增加和死亡。为了研究过表达人源E105D的表型,本研究首先观察了胚胎发育状态,发现和野生型相比,h或hE105D过表达并没有导致明显的发育异常(图3A)。然后,通过整胚原位杂交实验检测神经、肌肉特异性标记物,观察hE105D对神经、肌肉发育的影响。结果表明,与野生型和过表达h的胚胎相比,hE105D过表达胚胎中神经特异性标记基因的表达明显减少,表现为抑制性神经元标记基因、兴奋性神经元标记基因以及泛神经元标记基因的表达明显降低(图3B),说明hE105D影响神经发生。同时,本研究发现过表达hE105D导致肌肉特异性标记基因、以及的表达明显降低(图3C)。鬼笔环肽染色结果显示,hE105D过表达后导致胚胎肌丝宽度明显减小(图3D),进一步说明hE105D导致肌肉发育异常。
2.3 过表达人源TPI1E105D影响红系及髓系细胞发育
贫血是TPI DF的早期症状之一。为了检测本研究所构建的转基因品系是否影响红系发育,通过整胚原位杂交检测了红细胞相关标记基因的表达。结果显示,相比于野生型组和hWT过表达组,过表达hE105D导致24 hpf胚胎中原始红细胞生成关键因子、和的表达显著性减弱(图4A);而在2 dpf时,以及胚胎期珠蛋白基因的表达水平则明显增强(图4B)。临床研究报道,红细胞减少会使氧张力下降、红细胞生成素增加,进而促使红系造血代偿增加,并诱导未成熟的红细胞提前从骨髓释放[15]。因此,本研究推测2 dpf时红系相关基因的表达升高可能是由于早期红细胞减少后导致的红系代偿性增生。为了验证这一推测,通过吉姆萨染色(Giemsa staining)检测红系的发育状态,结果显示过表达hE105D导致红细胞的核质比明显增加(图4C),提示未成熟的红细胞增多。这些结果说明过表达hE105D初期会导致原始红细胞减少,随后引起红系代偿性增生。

图3 过表达人源TPI1E105D导致神经以及肌肉发育异常
A:转基因组斑马鱼明场下无明显发育异常。比例尺为100 μm;B:36 hpf时野生型组和转基因组神经相关标记基因的整胚原位杂交结果。其中是抑制性神经元的标记基因,是兴奋性神经元的标记基因,是泛神经元的标记基因,比例尺为100 μm;右侧为原位杂交胚胎数目统计结果,星号表示差异的显著性(****<0.0001,n.s.表示无显著性差异),值由卡方检验计算得出;C:36 hpf时野生型组和转基因组肌肉相关标记基因的整胚原位杂交结果。其中为体节发育相关的标记基因,为快肌纤维标记基因,为慢肌纤维标记基因,比例尺为100 μm;右侧为原位杂交胚胎数目的统计结果,星号表示差异的显著性(****<0.0001,n.s.表示无显著性差异),值由卡方检验计算得出;D:2 dpf时野生型组和转基因组鬼笔环肽染色。白色大方框放大自白色小方框,比例尺为20 μm;右侧为肌丝宽度统计结果,数据展示为平均值±标准差,星号表示差异的显著性(****<0.0001,n.s.表示无显著性差异),值由双尾检验计算得出。
在人体中,当红细胞减少速度超过红系代偿性增生速度时,就会发生贫血[16]。为了验证过表达hE105D是否最终会导致贫血表型,通过邻联茴香胺染色(-)检测野生型组及转基因品系胚胎中的血红蛋白水平,结果显示过表达hE105D胚胎中血红蛋白的含量明显减少(图4D);同时,通过流式细胞分析技术检测了红细胞数量,发现在hE105D过表达组中,:dsRed+红细胞数量显著减少(图4E)。这些结果说明在3 dpf时,过表达hE105D组中红细胞减少的速度已超过红系代偿性增生的速度,最终导致贫血。综上,过表达hE105D可导致血红蛋白含量减少和红系细胞数量减少,类似于临床上的贫血表型。
除了贫血外,TPI DF患者还存在免疫功能低下和易感染的临床症状。白细胞具有抵御有害微生物入侵的功能,比如单核巨噬细胞可以抵抗感染、吞噬异物,以及调节身体免疫力[17];中性粒细胞是抵御急性细菌感染的主要防线,其减少会增加感染风险[18]。为了初步了解hE105D是否导致免疫功能低下,本研究检测了髓系相关标记基因的表达,并观察单核巨噬细胞、中性粒细胞等白细胞的数量是否发生改变。分别是髓淋前体细胞、巨噬细胞和中性粒细胞的标记基因。在hE105D过表达的斑马鱼胚胎中,髓淋前体细胞、巨噬细胞以及中性粒细胞的数量均有所下降(图4F),提示hE105D过表达斑马鱼品系可能存在防御功能减弱,易于感染的表型。本研究同时检测了淋巴细胞以及造血干祖细胞。、为淋巴细胞的标记基因,为造血干祖细胞的标记基因。结果显示,与野生型和hWT过表达组相比,hE105D异常表达并没有影响淋系以及造血干祖细胞的发育(图4G)。

A:24 hpf时野生型组和转基因组红系生成相关基因的整胚原位杂交结果。下方为原位杂交统计结果;B:2 dpf时野生型组和转基因组红系相关基因的整胚原位杂交结果。下方为原位杂交统计结果。其中A和B小图中为红系前体的标记基因,为造血前体的标记基因,为胚胎期珠蛋白的标记基因,比例尺为100 μm;C:2 dpf时野生型组和转基因组吉姆萨染色结果。比例尺为25 μm;下方为红细胞核质比统计结果,每组纳入统计的红细胞数目均大于150个;D:3 dpf时野生型组和转基因组邻联茴香胺染色结果。比例尺为100 μm;右侧为染色面积的统计结果;E:3 dpf时野生型组和转基因组流式分析数据及统计图;F:2 dpf时野生型组和转基因组中髓系细胞相关基因的整胚原位杂交结果。其中为髓系淋系前体细胞的标记基因,为单核巨噬细胞的标记基因,为中性粒细胞的标记基因,比例尺为100 μm;下方为髓系细胞相关基因的整胚原位杂交统计结果;G:野生型组与转基因组中、和的整胚原位杂交结果。其中、为淋巴细胞的标记基因,为造血干祖细胞的标记基因,比例尺为100 μm;下方为原位杂交统计结果。数据展示为平均值±标准差,星号表示差异的显著性(*<0.05,**<0.01,***<0.001,****<0.0001,n.s.表示无显著性差异),值由双尾检验计算得出。
3 讨论
TPI DF是一种发生在儿童早期的罕见疾病,目前尚无有效的治疗方法。人类致病性突变可能通过影响TPI的功能域导致催化活性下降或蛋白稳定性降低,从而引起TPI DF。但也有研究指出,在不影响TPI蛋白质稳定性的情况下,其催化活性下降不足以引起TPI DF症状[19],因此该疾病的确切病因仍存在争议。通过对临床数据的分析发现,影响TPI二聚体形成的突变可能会引起更严重的症状[8],E105D可以影响TPI二聚体形成,除此之外它还是目前已报道病例中最常见的突变类型,这使得这一突变类型成为构建TPI DF疾病模型的首选位点。
有关TPI DF的动物模型已在小鼠()和果蝇()中建立,E105D/null小鼠存在神经肌肉功能障碍、溶血性贫血和寿命显著缩短的严重表型,是第一个具有神经肌肉表型的TPI DF小鼠模型[20]。TPI果蝇表现出进行性运动障碍、神经肌肉损伤和寿命缩短,研究人员通过TPI果蝇模型筛选鉴定了25种对TPI稳定性至关重要的蛋白质,这对于开发稳定TPI蛋白的药物用于治疗TPI DF具有参考意义[21]。斑马鱼作为一种脊椎模式动物,相比于无脊椎动物果蝇,在基因组和发育模式上都与人类有更高的相似性;相较于小鼠,斑马鱼具有发育繁殖能力强、饲养成本低、能进行高通量药物筛选的优势。因此,本研究构建的过表达hE105D转基因斑马鱼可以成为机制研究和药物筛选的新的模型,丰富研究者对于动物疾病模型的选择。
本研究借助转座子系统构建了稳定表达人源性E105D的Tg(:hE105D-eGFP)转基因斑马鱼,该转基因品系呈现出人类TPI DF相似的表型,主要表现为神经肌肉发育异常,伴有贫血、白细胞减少的血液系统表型,但是具体的发病机制尚不清楚。检测野生型组和转基因组中的TPI酶活性发现,过表达人源性E105D会导致斑马鱼整体的TPI酶活性降低(图2D),这可能是Tg(:hE105D-eGFP)转基因斑马鱼存在TPI DF的原因之一。除此之外,TPI的异常二聚化被认为在TPI DF中起关键作用,有研究认为TPI DF可能是蛋白构象疾病[5]。E105D突变靠近TPI二聚化结合位点,该突变会导致TPI活性二聚体形成障碍,而未形成二聚体的不稳定TPI单体具有高凝聚力,过多的TPI单体错误折叠形成有毒蛋白聚集体,从而引发神经功能障碍[5,6,22],我们推测这可能是人源性E105D能在斑马鱼体内存在磷酸丙糖异构酶的前提下仍能引起神经系统异常的可能原因。在检测人源性E105D异常表达的血液表型时,本研究发现红细胞以及髓系细胞均减少,而淋巴细胞的数量以及造血干祖细胞的产生并未明显改变(图4G),广泛表达的为何特异性的影响部分细胞类型?这可能是由于在不同细胞类型中,TPI的活性也存在差异[5],如在红细胞中,TPI突变蛋白可能与红细胞膜有更强的相互作用,导致TPI的微区室化,这会进一步降低TPI缺陷细胞的TPI活性[23]。
总之,这些数据表明,本研究所构建的Tg(:hE105D-eGFP)转基因品系具有与人类TPI DF类似的表型,包括神经、肌肉、血液系统在内的多方面表型,因此该转基因品系可以用于对TPI DF 进行多系统的机制研究,也为高通量药物筛选提供了一种合适的动物模型。
附加材料见文章电子版www.chinagene.cn。
[1] Schneider AS. Triosephosphate isomerase deficiency: historical perspectives and molecular aspects., 2000, 13(1): 119–140.
[2] Rieder SV, Rose IA. The mechanism of the triosephosphate isomerase reaction., 1959, 234(5): 1007–1010.
[3] Wierenga RK, Kapetaniou EG, Venkatesan R. Triosephosphate isomerase: a highly evolved biocatalyst., 2010, 67(23): 3961–3982.
[4] Schneider AS, Valentine WN, Hattori M, Heins HL. Hereditary hemolytic anemia with triosephosphate isomerase deficiency., 1965, 272: 229–235.
[5] Orosz F, Oláh J, Ovádi J. Triosephosphate isomerase deficiency: new insights into an enigmatic disease., 2009, 1792(12): 1168–1174.
[6] Orosz F, Oláh J, Ovádi J. Triosephosphate isomerase deficiency: facts and doubts., 2006, 58(12): 703–715.
[7] Daar IO, Artymiuk PJ, Phillips DC, Maquat LE. Human triose-phosphate isomerase deficiency: a single amino acid substitution results in a thermolabile enzyme., 1986, 83(20): 7903–7907.
[8] Schneider A, Cohen-Solal M. Hematologically important mutations: triosephosphate isomerase., 1996, 22(1): 82–84.
[9] Oliver C, Timson DJ.prediction of the effects of mutations in the human triose phosphate isomerase gene: towards a predictive framework for TPI deficiency., 2017, 60(6): 289–298.
[10] Ralser M, Heeren G, Breitenbach M, Lehrach H, Krobitsch S. Triose phosphate isomerase deficiency is caused by altered dimerization--not catalytic inactivity-- of the mutant enzymes., 2006, 1(1): e30.
[11] Zhang CX, Liu F. A brief protocol for high-resolution whole mounthybridization in zebrafish., 2013, 35(4): 522–528.张春霞, 刘峰. 斑马鱼高分辨率整胚原位杂交实验方法与流程. 遗传, 2013, 35(4): 522–528.
[12] Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis., 2012, 9(7): 676–682.
[13] Roland BP, Zeccola AM, Larsen SB, Amrich CG, Talsma AD, Stuchul KA, Heroux A, Levitan ES, Vandemark AP, Palladino MJ. Structural and genetic studies demonstrate neurologic dysfunction in triosephosphate isomerase deficiency is associated with impaired synaptic vesicle dynamics., 2016, 12(3): e1005941.
[14] Selamioğlu A, Karaca M, Balcı MC, Körbeyli HK, Durmuş A, Yıldız EP, Karaman S, Gökçay GF. Triosephosphate isomerase deficiency: E105D mutation in unrelated patients and review of the literature., 2023, 14(3): 231–238.
[15] Li XX, Li Y, Zhao X, Peng GX, Li JP, Ye L, Yang WR, Zhou K, Fan HH, Yang Y, Xiong YZ, Li Y, Song L, Jing LP, Zhang L, Zhang FK. Characteristics of bone marrow compensatory erythropoiesis in hereditary spherocytosis., 2022, 43(2): 115–119.李小霞, 李园, 赵馨, 彭广新, 李建平, 叶蕾, 杨文睿, 周康, 樊慧慧, 杨洋, 熊佑祯, 李洋, 宋琳, 井丽萍, 张莉, 张凤奎. 遗传性球形红细胞增多症骨髓红系造血代偿特征. 中华血液学杂志, 2022, 43(2): 115–119.
[16] Beguin Y, Clemons GK, Pootrakul P, Fillet G. Quantitative assessment of erythropoiesis and functional classification of anemia based on measurements of serum transferrin receptor and erythropoietin., 1993, 81(4): 1067–1076.
[17] Horwood NJ. Macrophage polarization and bone formation: a review., 2016, 51(1): 79–86.
[18] Segal AW. How neutrophils kill microbes., 2005, 23: 197–223.
[19] Segal J, Mülleder M, Krüger A, Adler T, Scholze-Wittler M, Becker L, Calzada-Wack J, Garrett L, Hölter SM, Rathkolb B, Rozman J, Racz I, Fischer R, Busch DH, Neff F, Klingenspor M, Klopstock T, Grüning NM, Michel S, Lukaszewska-Mcgreal B, Voigt I, Hartmann L, Timmermann B, Lehrach H, Wolf E, Wurst W, Gailus-Durner V, Fuchs H, de Angelis MH, Schrewe H, Yuneva M, Ralser M. Low catalytic activity is insufficient to induce disease pathology in triosephosphate isomerase deficiency., 2019, 42(5): 839–849.
[20] Myers TD, Ferguson C, Gliniak E, Homanics GE, Palladino MJ. Murine model of triosephosphate isomerase deficiency with anemia and severe neuromuscular dysfunction., 2022, 3: 100062.
[21] Hrizo SL, Eicher SL, Myers TD, Mcgrath I, Wodrich APK, Venkatesh H, Manjooran D, Swoger S, Gagnon K, Bruskin M, Lebedev MV, Zheng S, Vitantonio A, Kim S, Lamb ZJ, Vogt A, Ruzhnikov MRZ, Palladino MJ. Identification of protein quality control regulators using amodel of TPI deficiency., 2021, 152: 105299.
[22] Mainfroid V, Terpstra P, Beauregard M, Frère JM, Mande SC, Hol WG, Martial JA, Goraj K. Three hTIM mutants that provide new insights on why TIM is a dimer., 1996, 257(2): 441–456.
[23] Orosz F, Wágner G, Liliom K, Kovács J, Baróti K, Horányi M, Farkas T, Hollán S, Ovádi J. Enhanced association of mutant triosephosphate isomerase to red cell membranes and to brain microtubules., 2000, 97(3): 1026–1031.
Generation and analysis of TPI deficiency zebrafish model
Piao Sun1, Ying Li1, Fan Liu1, Lu Wang1,2
Triosephosphate isomerase deficiency (TPI DF) is a severe multisystem degenerative disease, manifested clinically as hemolytic anemia, neuromuscular abnormalities, and susceptibility to infection, frequently leading to death within 5 years of onset. There is a lack of effective clinical treatment as the pathogenesis underlying TPI DF remains largely unknown. In this study, we generate a transgenic zebrafish line [Tg(:E105D-eGFP)] with the humanE105D(hE105D) mutation, which is the most recurrent mutation in TPI DF patients. Overexpression of hE105Daffects the development of erythroid and myeloid cells and leads to impaired neural and muscular development. In conclusion, we create a TPI DF zebrafish modelto recapitulate the majority clinical features of TPI DF patients, providing a new animal model for pathogenesis study and drug screening of TPI DF.
triosephosphate isomerase deficiency (TPI DF);E105D; zebrafish;disease model
2023-12-22;
2024-02-05;
2024-02-22
国家自然科学基金项目(编号:32222027, 32170838)和天津市杰出青年项目(编号:21JCJQJC00120)资助[Supported by the National Natural Science Foundation of China (Nos.32222027, 32170838) and the Science Fund for Distinguished Young Scholars of Tianjin Municipality (No. 21JCJQJC00120)]
孙飘,硕士研究生,专业方向:干细胞与再生医学。E-mail: sunpiao@ihcams.ac.cn
李颖,博士,研究方向:干细胞与再生医学。E-mail: liying3@ihcams.ac.cn
孙飘和李颖同为第一作者。
王璐,博士,研究员,研究方向:发育生物学,干细胞与再生医学。E-mail: wanglu1@ihcams.ac.cn
10.16288/j.yczz.23-316
(责任编委: 张文清)
猜你喜欢
学苑创造·A版(2023年6期)2023-06-16 00:06:35
基础医学与临床(2022年7期)2022-07-11 02:39:10
基础医学与临床(2022年6期)2022-06-27 01:59:08
小天使·二年级语数英综合(2021年8期)2021-08-16 10:56:50
中成药(2017年6期)2017-06-13 07:30:35
特产研究(2016年3期)2016-04-12 07:16:32
基础医学与临床(2015年6期)2015-07-31 22:49:40
中国当代医药(2015年17期)2015-03-01 02:03:43
郑州大学学报(医学版)(2015年1期)2015-02-27 14:50:35
郑州大学学报(医学版)(2015年1期)2015-02-27 14:50:35
