
细胞焦亡在骨代谢异常疾病中的研究
2024-03-13 06:20杨小瑞曹林忠胡康一张勇杰尚征亚万超超
中国骨质疏松杂志 2024年1期
杨小瑞 曹林忠,2* 胡康一 张勇杰 尚征亚 万超超
1甘肃中医药大学,甘肃 兰州 730000
2甘肃中医药大学附属医院,甘肃 兰州 730099
骨代谢相关细胞在骨组织的合成、分泌、吸收、矿化等过程中发挥着重要作用,是骨修复重建的关键细胞。骨代谢相关细胞分化及功能的异常与许多骨代谢疾病的发病机制密切相关。
随着对细胞焦亡与骨代谢细胞相关研究的不断深入,细胞焦亡过程中炎症小体的激活及下游关键细胞因子的表达在骨代谢过程中具有重要调控作用,会导致骨代谢调节处于紊乱状态,打破骨代谢过程中骨代谢相关细胞骨形成与骨破坏作用的动态平衡,是骨代谢异常疾病发生发展的重要病理环节。本文就细胞焦亡在骨代谢相关细胞中发生的分子作用机制与激素性股骨头坏死、类风湿关节炎、骨质疏松症、骨关节炎等骨代谢异常疾病的相关研究进行综述,以期为进一步探究骨代谢异常疾病的预防和治疗提供理论参考和指导。
1 细胞焦亡概述
细胞焦亡(pyroptosis)又称促炎性程序性细胞死亡,1986年Friedlander 等[1]最早在原代小鼠腹腔巨噬细胞中发现该自然死亡现象并首次提出细胞焦亡的概念。细胞焦亡是由打孔蛋白Gasdermin-D介导核苷酸结合寡聚化结构域样受体蛋白3 (nucleotide-binding oligomerization domain-like receptor protein 3,NLRP3)炎症小体参与并依赖半胱氨酸天冬氨酸蛋白酶(主要是caspase-1,4,5,11)的新型促炎症细胞程序性死亡方式[2],包括依赖caspase-1和非依赖caspase-1两种途径(见图1),常发生在单核细胞、巨噬细胞和树突状细胞等一些专业吞噬细胞中[3]。其形态学特征表现为细胞发生肿胀,形成焦亡小体,细胞膜破裂,释放炎症因子[4],是机体抵抗外界有害因素刺激的重要的天然免疫反应之一。在某些病理因素(如氧化应激、高血糖、炎症损伤等)的作用下,细胞焦亡出现过度激活,导致骨代谢相关细胞发生死亡并介导多种骨代谢异常疾病的发生[5]。
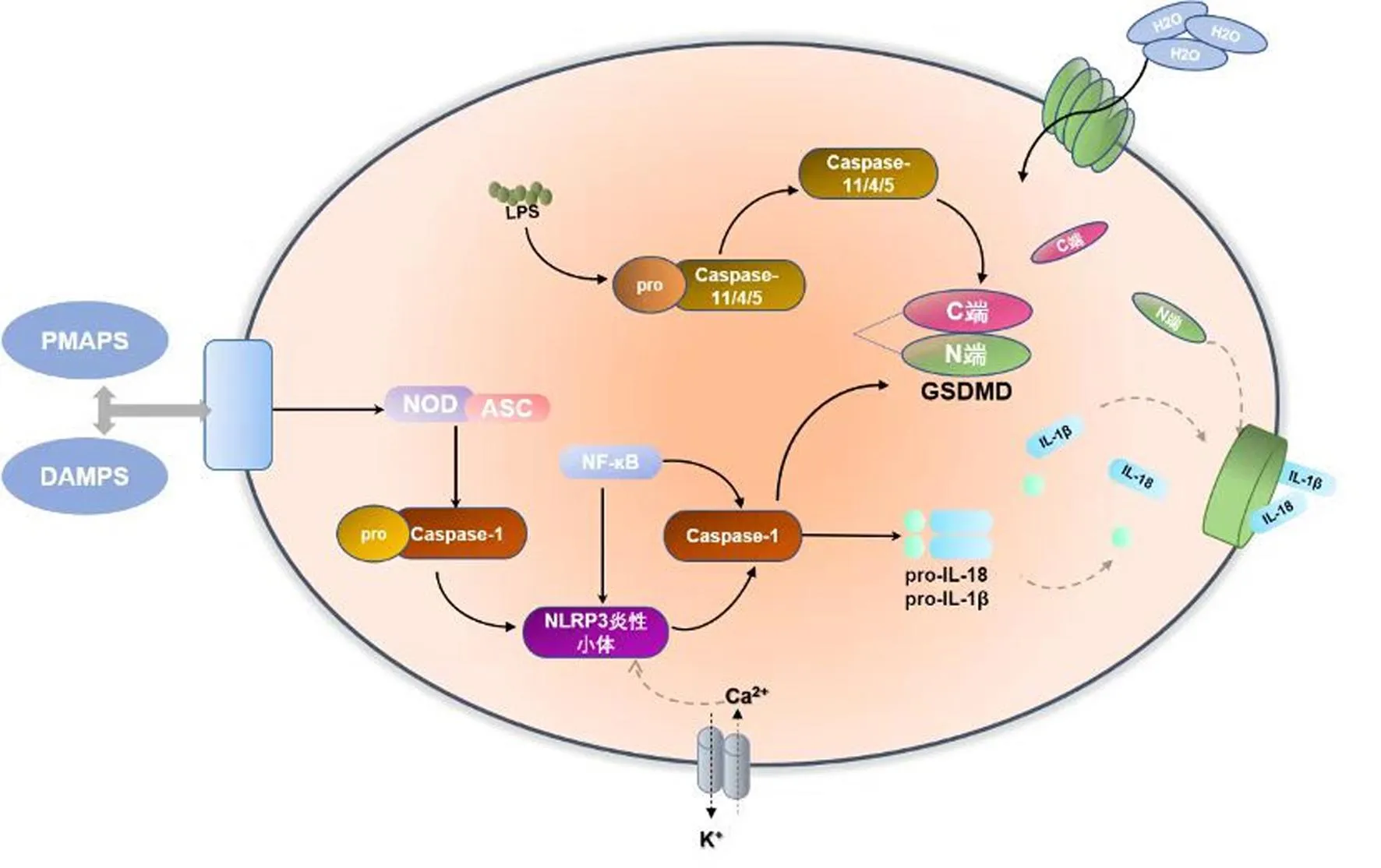
图1 细胞焦亡经典和非经典途径发生机制
2 细胞焦亡与骨代谢异常疾病
2.1 细胞焦亡与激素性股骨头坏死
激素性股骨头坏死(steroid-induced avascular necrosis of the femoral head,SANFH)是由于糖皮质激素(glucocorticoids,GC)过量使用致使股骨头内微循环障碍、骨吸收/形成代谢失衡,出现以股骨头局部组织无菌性炎症、变性、坏死、塌陷为主要病理表现的骨代谢性疾病[6]。
GC属于类固醇类,常用于感染性、自身免疫性疾病。在长期使用过程中发现,GC会影响脂质代谢,造成脂肪堆积,形成的脂肪栓子会导致骨代谢紊乱和炎症反应上调[7]。炎症可以抑制BMSCs 向成骨细胞、成脂细胞或成软骨细胞的分化。NLRP3及相关细胞因子作为启动细胞焦亡的重要免疫信号复合物,贯穿细胞焦亡引起的炎症反应全过程,对SANFH骨代谢和稳态的调节具有重要作用[8]。高浓度GC暴露会破坏PC-12细胞内质网内稳态导致内质网中未折叠或错误折叠的蛋白质积累,通过激活内质网应激可以诱导NLRP3炎症小体的活性增强,显著增加炎症小体的数量[9],炎性小体的过度激活会通过调节细胞因子(如T辅助细胞17)来诱导骨溶解,导致破骨细胞活性增加,成骨细胞活性降低。在GC诱导的大鼠SANFH模型中,GC通过刺激线粒体产生应激,刺激活性氧(ROS)大量增加,核因子-κB(NF-κB)信号通路被激活,使得破骨细胞活性及相关蛋白的表达显著上调,造成股骨头骨量大量丢失[10]。
Jiang等[11]在体外高糖(HG)诱导大鼠BMSCs糖尿病模型发现,HG可能会刺激TXNIP的表达,TXNIP与硫氧还蛋白(TRX)分离,通过TXNIP/NLRP3信号通路在胞内的转导,可以直接结合并激活NLRP3炎症小体各组分,并促进TNF-α和IL-1β、IL-18等炎症因子的高度表达,导致BMSCs炎症反应的发生,从而抑制BMSCs成骨分化的能力[12]。Yang等[13]在构建的体外体内糖尿病模型牙槽骨内检测发现成骨细胞焦亡相关蛋白caspase-1、GSDMD的表达水平明显升高,并且使用caspase-1抑制剂可以降低对成骨细胞增殖分化的抑制作用。Rocha等[14]通过体外构建LPS诱导小鼠破骨细胞发生牙周炎的模型中分别敲除小鼠NLRP3基因和caspase-1基因后,小鼠破骨细胞的骨吸收程度都被削弱且破骨细胞的上游信号分子NF-KB发生转导,导致促炎因子的数量增加。Drummer等[15]发现早期高脂血症可以诱导caspase-11激活,活化后释放的大量促炎症细胞因子IL-18参与并调控高脂血症诱导的体内血管内皮细胞死亡相关基因的表达,同时介导BMSCs血管内皮细胞炎症反应的发生[16],加重坏死股骨头局部缺血缺氧状态。
2.2 细胞焦亡与骨质疏松症
骨质疏松症(osteoporosis,OP)是一种全身性的临床常见代谢性骨病,通常好发于中老年人群,主要由于骨形成/骨吸收失衡所致,包括原发性、继发性和特发性三种类型,其特点是骨密度降低、骨量减少和骨组织微观结构改变,致使骨的脆性增加,常易引起脊柱变形、骨折等并发症,并增加死亡风险[17]。
NLRP3炎症小体是一种细胞内多蛋白复合物,由NOD样受体(NOD-like receptor)、凋亡相关斑点样蛋白(apoptosis-associated speck-like protein,ASC)、pro-caspases组成,具有调节机体固有免疫的作用。NLRP3炎症小体的激活依赖上游丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPKs)、NF-κB信号通路以及有效激动剂如离子转移、ROS等重要因素[18]。衰老、糖代谢紊乱、激素的使用以及绝经后雌激素缺乏可以促使NF-κB活化,刺激骨微环境中慢性炎症水平升高,诱导NLRP3和IL-1β前体蛋白过表达,NLRP3炎症小体的异常激活不仅能够通过促进炎症因子IL-1β和IL-18的产生来介导破骨细胞的分化,还可以通过上调Caspase-1和GSDMD的表达造成成骨细胞破裂死亡和功能障碍[19-20]。
An等[21]研究发现,在高糖(high glucose,HG)诱导的体外破骨细胞(OCs)中,HG 可以诱导ROS的产生以及MAPKs、NF-κB信号通路相关蛋白的表达,通过ROS/MAPKs/NF-κB/NLRP3信号通路的转导不仅可以促进NLRP3炎症小体的激活,还可以在NF-κB的活化作用下扩大炎症反应从而增强破骨细胞的骨吸收作用。罗立慧等[22]采用HG处理小鼠胚胎成骨细胞MC3T3-E1可明显促进NLRP3和caspase-1基因的表达且会引起成骨细胞MC3T3-E1发生肿胀破裂,诱导细胞焦亡的发生,同时造成成骨细胞炎症和损伤的发生,导致成骨细胞分化与矿化功能下降,骨生成作用减弱。鸢尾素作为一种可以抗衰老并且可以强化骨骼的骨骼肌分泌物,与衰老或绝经后骨质疏松密切相关[23]。Morgan等[24]发现经鸢尾素干预后的卵巢切除(OVX)大鼠,可检测出抗氧化因子Nrf2蛋白以及Runx2成骨基因的mRNA的表达水平明显下降,而NLRP3、Caspase-1的表达与前者正好相反,这也进一步证明衰老或雌激素缺乏引起的慢性炎症微环境是促使细胞发生焦亡的关键因素。
2.3 细胞焦亡与骨关节炎
骨关节炎 (osteoarthritis,OA) 是一种由于骨自主修复和破坏之间的不平衡引起的慢性炎症性关节疾病。软骨基质降解、滑膜炎症和软骨下骨重塑等是OA病变过程中的典型病理表现[25]。软骨细胞异常肥大或死亡导致软骨细胞代谢失衡从而出现软骨基质降解及软骨骨质破坏[26][27]及滑膜组织中具有高代谢活性的巨噬样滑膜细胞吞噬浸润作用介导的滑膜炎症反应和成纤维样细胞的纤维化作用引起的细胞外基质沉积均是 OA 发病的关键触发因素[28-29]。
研究认为关节急性损伤、慢性劳损、关节异物或异常的生物应力导致ROS和炎症因子的累积和释放与OA的发生发展关系密切[30]。ROS是体内一类氧的单电子还原产物,高水平的ROS会引起线粒体膜去极化,造成线粒体途径的ROS大量增多,ROS过度激活诱导NLRP3激活和GSDMD氧化,导致软骨细胞发生焦亡而肿胀破裂死亡,促进软骨退行性改变的发生[31]。同时,作为炎症介质的IL-1β和IL-18 已明确证实可以促进软骨细胞死亡,并通过抑制Ⅱ型胶原蛋白和蛋白聚糖的产生来加速软骨细胞外基质的降解,从而导致OA的发生发展[32]。
Bostan等[33]利用LPS/ATP诱导构建体外滑膜成纤维细胞焦亡模型,发现NLRP3炎症小体在OA 患者膝关节滑膜组织中呈现高表达状态;敲除NLRP3基因后能够显著降低下游信号分子ASC、caspase-1、GSDMD基因和蛋白的表达水平,可以减缓滑膜成纤维细胞焦亡的发生。智佳佳等[34]发现高浓度的LPS/ATP可以诱导C28软骨细胞内ROS大量产生从而激活NLRP3炎症小体,明显降低软骨细胞活性。另外,研究发现钙化软骨区软骨下骨血流灌注减少会引起软骨下骨缺氧,可以刺激低氧诱导因子-1α (hypoxia-inducible factor 1-alpha,HIF-1α)的表达。HIF-1α可以促进促炎因子和生长因子的产生,从而激活成纤维细胞并参与 OA 滑膜纤维化病理过程的形成[35];同时又可以通过诱导NLRP3炎症小体活化促进滑膜成纤维细胞发生焦亡[36],明确证实细胞焦亡在OA 软骨和滑膜细胞病变过程中发挥着重要的调控作用。
2.4 细胞焦亡与类风湿关节炎
类风湿关节炎(rheumatoid arthritis,RA)是发生在滑膜内衬及软骨表面并累及周围关节的一种常见的慢性、炎症性、自身免疫性疾病。RA的基本病理特征为滑膜炎症和骨与软骨组织的破坏[37]。炎症作为先天免疫的主要反应,滑膜内炎症细胞浸润是RA发生的重要驱动因素。
细胞焦亡是一种新型促炎性细胞死亡形式,在RA和NLRP3基因遗传多态性分析中,研究发现NLRP3 rs10754558 C/G多态性与中国汉族RA患者发病风险、发病人群和临床特征密切相关[38]。NLRP3炎症小体是连接先天免疫和适应性免疫的重要桥梁[39],活化后的NLRP3炎症小体可以激活caspase-1和GSDMD,产生具有生物活性的促炎细胞因子IL-1β和IL-18。IL-1β和IL-18是IL-1家族中促进先天免疫系统细胞活化的重要炎症调节因子,在RA的病理过程中发挥着关键作用[40]。IL-1β是RA的主要致病因素之一,可以上调破骨细胞前体细胞上的NF-kB受体激活因子(RANK)的表达,使RANK与核因子kB受体激活剂配体(RANKL)相结合[41-42],从而增强其活性并刺激破骨细胞的产生以诱导骨与软骨组织的破坏[43]。IL-1β也可以与其他促炎症因子如TNF-a发生协同作用[44],进一步放大炎症反应并加速骨丢失。IL-18是一种多效性细胞因子,在RA炎症反应的级联上调和维持中发挥重要作用。IL-18通过激活滑膜中的T细胞产生炎性细胞因子、RANKL等[45],介导骨破坏的发生。
王秋苑等[40]发现RA患者血清和关节液中的NLRP3、caspase-1、IL-1β、IL-18表达水平明显升高,且与疾病活动程度和滑膜炎性水平呈正相关,证实细胞焦亡相关因此参与了滑膜细胞焦亡导致的RA关节滑膜炎的病理过程。DNA聚合酶β (DNA polymerase β,Pol β)是一种碱基切除修复的关键酶,对RA稳定器损伤的骨组织具有修复作用。Gu等[46]研究报道在活动性RA患者和胶原诱导性关节炎小鼠的外周血单核细胞中,Pol β的表达显著降低,而在RA巨噬细胞焦亡模型中,Pol β敲除会导致DNA损伤积累,诱导NF-kB信号通路转导并上调NLRP3、IL-1β和IL-18的表达。因此,靶向NF-kB-NLRP3对于改善促炎细胞因子造成的炎症损伤具有重要意义。
3 小结与展望
细胞焦亡作为一种新型的促炎性程序性死亡,关于焦亡相关细胞因子及信号通路与骨代谢异常疾病发病机制的相关性已经得到了初步研究证实。NLRP3炎症小体作为细胞焦亡发生的关键功能分子,在骨代谢相关细胞内可以通过NF-κB信号通路转导、氧化应激、线粒体功能障碍等多种途径被激活介导炎症反应的发生;同时由NLRP3炎症小体介导的IL-1β和IL-18又可以通过RANK、MAPKs等细胞因子的调节影响骨代谢相关细胞的增殖、分化、吸收等功能,造成骨代谢紊乱及骨稳态失衡,导致骨代谢异常疾病的发生发展。
本文通过系统综述细胞焦亡过程中炎症小体的激活以及下游促炎症因子IL-1β和IL-18成熟对骨代谢相关细胞的影响以及与激素性股骨头坏死、类风湿关节炎、骨质疏松症、骨关节炎等发病机制的相关性,证实细胞焦亡在骨代谢异常疾病中发挥着重要调控作用。但是除此之外,关于细胞焦亡还有更多值得我们去探索和挖掘的热点,比如人体免疫调节系统以及细胞内环境稳态对细胞焦亡是否具有分级靶向的作用以及如何有效避免焦亡过度激活对骨与关节组织造成严重损伤。相信随着未来对细胞焦亡不断地深入研究,可以进一步揭示关键信号通路和分子作用靶点,为阐明细胞焦亡与骨代谢疾病的发病机制研究及防治策略奠定基础,并为临床研究改善骨代谢异常疾病的新型靶向抑制剂药物提供理论支撑和指导,将其更好地运用于未来临床防治,从而有效减缓骨代谢异常疾病的发展进程,有效提高患者临床生存质量及预后。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
医学综述(2022年7期)2022-04-19
昆明医科大学学报(2020年12期)2021-01-26
世界科学技术-中医药现代化(2020年2期)2020-07-25
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
安徽医科大学学报(2016年12期)2017-01-15
中国民族医药杂志(2016年6期)2016-05-09
西南军医(2015年3期)2015-04-23
中国医科大学学报(2015年10期)2015-03-01
国际心血管病杂志(2015年5期)2015-02-27
