
N6-腺苷酸甲基化干预细胞增殖分化在神经系统疾病中的进展
2024-03-13 11:08:58候雅清张雷雷冯香瑞马晶莹马旺然常翔杨琳
河北医药 2024年2期
候雅清 张雷雷 冯香瑞 马晶莹 马旺然 常翔 杨琳
N6-腺苷酸甲基化(N6-methyladenosine,m6A)通过重塑RNA的二级结构来改变其翻译后的功能。甲基化转移酶将供体S-腺苷甲硫氨酸(SAM)上的甲基催化转移至RNA腺嘌呤的第6个N元素上;去甲基转移酶擦除该修饰,使得该过程具有动态可逆性;通过阅读蛋白特异性识别修饰信号后发挥生物学作用[1]。迄今为止,生物体中已被鉴定的RNA修饰方式高达170多种[2],例如N6-甲基腺嘌呤(N6-methyladenosine,m6A),N6,2-O-二甲基腺嘌呤核苷(N6-2-dimethyladenosine,m6Am)以及假尿苷(Pseudouridine,ψ)[3]等,其中m6A约占RNA甲基化修饰的80%,并广泛分布于各型RNA[4]。作为影响mRNA的“生命周期”的核心因子,在细胞增殖、分化、代谢、应激反应等生命活动发挥重要的作用,这种化学修饰调节人类发育、健康和疾病[5]。而在神经系统中,m6A水平升高与脑组织发育成熟高度相关,研究发现被抑制METTL14的小鼠胚胎模型中的NSCs自我更新与增殖分化同样依赖于m6A修饰[6]。m6A水平失衡在中枢神经系统相关疾病中的重要性不容忽视,因此,本文综述m6A甲基化干预细胞增殖分化在神经系统疾病中的作用,为精确干预病程和预后提供新的治疗靶点。
1 m6A修饰的动态调控机制
m6A主要沉积在含有特殊序列RRACH(D=r=A/G/U,r=A/G,H=A/C/U)的终止密码子、以及非翻译区(UTR)的3’端[7]。m6A修饰的调节因子共同作用参与RNA的β折叠过程[8],维持RNA上m6A沉积的动态稳定并参与生物体中枢神经系统中更复杂的生理病理机制,参与修饰的相关蛋白见表1。
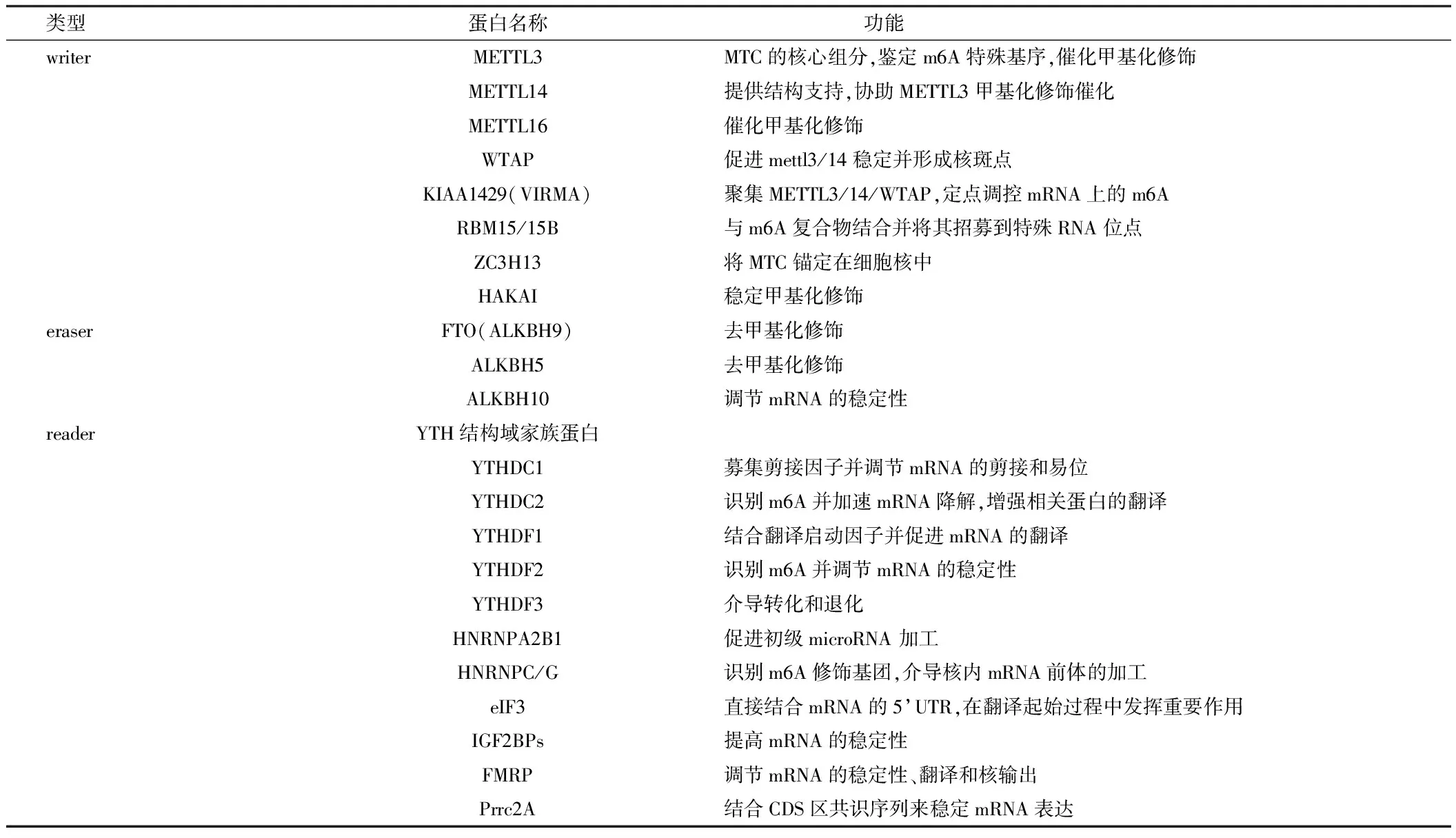
表1 m6A修饰相关蛋白的功能
编码蛋白“Writers”通过在目标靶点上安装甲基来实现甲基化,该过程是由甲基转移酶复合物(MTC)操控,主要由METTL3/14(异源二聚体)、WTAP、KIAA1429、ZC3H13、RBM15/15B等构成。METTL3通过SAM结合域和一个DPPW基序(Asn-Pro-Pro-Trp)实现m6A的沉积[9];METTL14协助底物RNA识别。调控亚基WTAP协助稳定异源二聚体定位并与RBM15、ZC3H13、VIRMA相关转录因子结合形成一种新的蛋白复合物,精确定位靶点并参与m6A修饰的产生[10]。敲除ZC3H13导致WTAP、HAKAI和VIRMA发生易位,m6A水平显著降低60%~70%,影响细胞的自我更新[11]。
消码蛋白“Erasers”通过去除m6A修饰来实现甲基化逆转。常见的RNA去甲基化酶 有ALKB 家族的肥胖相关蛋白(FTO)和ALKB同源蛋白5(ALKBH5)[12]。2011年Wei等[13]发现Exportin2蛋白引导FTO在细胞质和细胞核之间穿梭,通过在细胞中的定位来决定它对底物的偏好;敲除FTO导致m6A在mRNA中的数量增加,首次证明了FTO的去甲基效应。ALKBH5是第2个被发现的去甲基化酶,其定位在细胞核中,仅用于前体mRNA,并表现出去甲基化的能力。在HeLa细胞中,ALKBH5下调使总mRNA中m6A含量增加[14]。尽管FTO与ALKBH5为同族成员,二者以不同的机制清除甲基,分别发挥着不同的生物作用。
阅读蛋白“Readers”利用特殊结构域识别m6A修饰后的碱基,从而使转录后调控具有真正意义上的生物学功能,常见的阅读蛋白主要包括含YTH家族结合蛋白、IGF2BPs、eIF3、HNRNP等,糖皮质激素受体GR作为RNA结合蛋白和转录因子,也可结合m6A[15]。YTHDCs和YTHDFs在细胞中的定位不同,在转录产物的运输过程及代谢中发挥不同作用。有研究发现IGF2BP家族通过KH结构域特异性地识别RNA上m6A修饰的UGGAC序列[16],并促进其翻译和稳定性。这些隐藏在结合蛋白中特殊结构域的发现有助于理解m6A在生理病理中的作用。
2 m6A生物学功能
m6A修饰参与mRNA代谢的整个过程,如miRNA成熟体加工、可变剪接、核运输、翻译和稳定性等过程中发挥不可替代的作用[17]。通过调控基因的表达或结构,在各种生物过程中起着关键作用,包括组织发育、氧化应激、热休克反应和DNA损伤反应,也可调控细胞的多种表型,如精原细胞分化和减数分裂的起始,影响造血干细胞(HSC)分化的命运,同样m6A在神经细胞中也起着至关重要的作用[18]。
3 m6A在神经发育中的作用
3.1 m6A与脑组织发育 随着大脑逐渐发育成熟,m6A修饰也相应增加,在敲低METTL3的小鼠神经系统中发现,小脑严重萎缩[19]。此外,ALKBH5在低压低氧环境中存在缺陷,导致小脑发育相关基因m6A修饰紊乱,进一步表明m6A修饰与脑组织发育密切相关。FTO在神经发生晚期表达最高,ALKBH5表达在大脑发育过程中下降,这可能意味着去甲基化酶对神经发生的过程发挥调节作用。而不同脑区域中显示m6A修饰水平有明显差异,如小鼠小脑中m6A修饰浓度高于大脑皮层[20],这些结果表明甲基化水平的差异均可能影响脑组织的生物学功能。
3.2 m6A调控神经细胞 研究表明,m6A含量的动态稳定在神经细胞功能和活动中至关重要,Meyer等[6,21]在成人大脑中m6A可调控神经干细胞分化。YTHDF1促进m6A甲基化mRNA翻译,刺激神经元促进学习记忆。Wang等[22]发现m6A修饰相关蛋白的失调与中枢神经系统发生有关联。研究发现,FTO缺失导致海马下颗粒区(SGZ)和侧脑室下脑室区(SVZ)成体干细胞数量显著下降,并损害成长阶段神经元的分化能力[23]。而在小鼠胚胎模型中发现,低水平METTL14破坏皮质发育,导致小鼠过早死亡;METTL3缺失抑制了神经元发育成熟。另有研究中,果蝇神经元中缺失METTL3与YTHDF1/2/3任何一种,都会对其短期记忆造成严重损害[24]。YTHDF2参与小鼠皮质神经的发育,敲低水平后使得神经元分化受损,最终导致小鼠大脑皮层发育延迟和功能障碍。
4 在神经系统疾病中
m6A修饰调节人类发育、健康和疾病,由于在大脑中的含量较高,表明它脑血管和其他神经疾病中的重要性不容忽视[25]。异常水平的m6A与CNS疾病的发生发展以及预后相关,下文重点阐述m6A修饰及相关蛋白介导基因干预细胞增殖分化对CNS疾病的影响,并为疾病的预防提供治疗靶点。
4.1 缺血性脑损伤 缺血性脑卒中(Ischemic stroke,IS)的致残致死性对患者的生活及寿命造成了严重的损害。病理机制大致为缺血-再灌注(I-R)损伤导致广泛的神经血管损伤。为了迅速介导卒中后的血管修复,需探索新的治疗思路。
有研究表明,m6A为新的IS的治疗靶点和诊断生物标志物,通过基因干预NSCs参与IS的发生和发展及预后[26]。Xu等[27]在大鼠模型中发现,大脑中动脉闭塞后的皮层和I-R后的初级神经元中,m6A水平随着ALKBH5的升高和FTO表达的下降而连续升高,敲除ALKBH5会加重神经元损伤。Chokkalla等[28]观察局灶缺血神经元发现,FTO的表达在mRNA和蛋白水平上都被下调,这可能导致卒中后大脑中m6A标记RNA的去甲基化降低,同时发现总m6A去甲基酶活性的下降。Li等[29]在大鼠模型中发现,敲低YTHDC1可加重缺血性脑损伤,过表达YTHDC1促进RNA降解,增加Akt磷酸化,从而促进神经元存活,尤其是保护缺血后脑损伤。内源性NSCs应答脑损伤时,分泌神经营养因子等可以刺激NSCs损伤后的增殖和分化,从而促进早期缺血性中风的内源性修复机制。相较NSCs移植治疗缺血性脑损伤,靶向调控m6A相关蛋白,间接调节NSCs的分化,避免了移植过程中的问题。
m6A甲基化的研究将为理解IS机制提供新的见解,可能成为IS的一个前瞻性治疗靶点。但m6A在IS中的具体作用机制还有待进一步探讨。
4.2 阿尔茨海默病(Alzheimer’s disease,AD) AD是一种以病理性大脑皮层萎缩为典型特征的不可逆的神经系统变性疾病。发病机制尚未完全确定,目前认为细胞外Aβ淀粉样蛋白沉积和细胞内τ蛋白过度磷酸化导致神经纤维缠绕[30]。
研究发现高浓度FTO通过激活雷帕霉素靶蛋白(target of rapamycin,mTOR)信号通路,加快神经元τ蛋白磷酸化[29]。Huang等[31]在AD患者死后的脑样本中发现高浓度的METTL3,推测AD患者海马内异常表达的METTL3可能参与了与AD的发病;而后在研究AD患者mRNA水平时发现RBM15B含量增加,这一结果可能说明m6A相关酶含量下降后的代偿反应。Han等[32]对比APP/PS1双转基因小鼠与正常小鼠的大脑皮质和海马区m6A的含量,结果发现模型组表达较高,其中Mettl3高表达而FTO表达降低;进一步测序分析基因发现,AD模型小鼠与正常小鼠参与突触生长的基因AMPA和NMDA基因甲基化水平升高,而SEMA基因甲基化水平明显降低。
尽管AD的具体机制仍未明确,但至少证明m6A可能参与了AD的病理过程。目前无论是逆转AD病理产物的药物设计,还是靶向淀粉样蛋白或τ蛋白的免疫疗法,临床试验未证明疗效显著的方案。从理论上讲,使用基因靶向调控NSCs来恢复受损的胆碱能神经元和大脑连接,可能会为AD提供新的治疗选择。然而,在临床使用前,仍然存在需要克服的障碍。
4.3 帕金森病 帕金森氏病(Parkinson’s disease,PD) PD是一种常见的年龄相关性神经退行性疾病。目前大多认为,发病过程与α-突触核蛋白(α-synuclein)过度聚集和纹状体黑质DA坏死相关, 近年来,m6A与PD病理机制的联系成为了研究热点。
m6A维持正常成年小鼠纹状体功能和学习能力,而在PD模型中,大脑纹状体区m6A水平明显降低,导致DA表达降低;敲低METTL14不改变小鼠纹状体大小,但可提高神经元兴奋性。Koranda等[33]发现,纹状体中METTL14的缺乏增加了对DA药物的敏感性,如D1R激动剂SKF-81297。研究发现PD患者纹状体区域m6A水平的明显降低与FTO浓度有关,认为FTO过表达加重线粒体损伤和多巴胺能神经元细胞损伤[34]。Chen等[35]在神经毒素6-羟多巴胺诱导的PC12(大鼠肾上腺嗜铬细胞瘤克隆的分泌DA的细胞系)细胞和PD大鼠脑纹状体时发现,抑制多巴胺能细胞内m6A水平,可诱导N-甲基-d-天冬氨酸受体1的表达,促进氧化应激和Ca2+内流,最终导致DA凋亡。而在药理研究中发现,可卡因可能通过降低FTO的表达来调节突触的成熟和定位,抑制DA再摄取来放大DA信号,缓解临床症状[34]。
目前PD病患者的治疗主要包括多巴胺类药物代替治疗、肉毒素治疗及脑深部电刺激术等,均不能阻止疾病发生发展,若利用m6A与这些受体的联系,或干预产生正常功能的DA,可能开发出增加受体数量、促进DA在脑内扩散的相关药物来治疗PD。
4.4 胶质母细胞瘤(Glioblastoma Multiforme,GBM) GBM是一种神经胶质细胞来源恶性肿瘤,患者常表现为头痛、记忆力减退或思维混乱等。研究发现,胶质母细胞瘤干细胞(GSCs)选择性地依赖于特殊结合位点促进肿瘤代谢,因此,靶向GSCs可能改善肿瘤结局[36]。
Huff等[37]认为m6A修饰中任何部分的失调都与多种癌症的不良预后和肿瘤发生有关;Gao等[38]在实验中无论抑制METTL3/14或FTO/ALKBH5过表达都可以加快GSCs自我更新并促进肿瘤的生长。Visvanathan等[39]发现METTL3过表达后m6A水平上升,损害了体外多个GSC系的肿瘤增殖。Zhang等[40]研究阐明m6A甲基化的缺失促进了体外和体内肿瘤的生长,敲低GSC来源的小鼠中的ALKBH5可以阻碍肿瘤恶性增殖。还有研究显示,YTHDF2或YTHDFs可能对癌基因MYC表达和肿瘤生长抑制产生更强的影响,可作为GBM中干扰MYC信号通路的一个广泛治疗靶点[41-42]。
尽管近年来针对GBM有很多新颖治疗方案,但由于95%的疗法仍无法穿透血脑屏障使得治疗几乎没有进展。通过靶点调控m6A相关蛋白水平精准阻止GSC的恶性增殖,或植入“清除因子”对恶意增殖的癌细胞进行定向清除。
4.5 重度抑郁症(major depression disorder,MDD) MDD是一种复发风险高的严重精神疾病,发作期存在明显的情感、认知和躯体症状,发作间期症状缓解。尽管MDD的发病机制尚不清楚,多认为数个影响基因表达的效应变异结合与环境等因素相互作用,可能导致MDD的病理进展。控制基因表达的m6A甲基化,以一种大脑区域和基因特异性的方式对环境刺激做出动态反应,其调节异常可能导致压力相关疾病,MDD中许多失调的系统被发现是通过m6A介导的,而越来越多的证据表明FTO在抑郁症病理中存在失调[43-45]。因此,这可能对理解MDD的分子机制有重要意义。MDD与情绪调节和认知功能相关的大脑区域内和跨区域的神经元通信中断有关。Liu等[43]研究发现MDD患者和抑郁小鼠模型海马区FTO含量下降,继续分析3种抑郁症小鼠模型中(慢性不可预测轻度应激(UCMS)、慢性克制应激(CRS)和社会失败应激(SDS)模型)m6A修饰酶的表达,UCMS小鼠模型中FTO和ALBKH5的mRNA表达较对照组明显下调,而RNA甲基转移酶的表达水平保持不变,并检测到多个情绪调节相关区域mRNA表达明显活跃。Samaan等[44]报道了FTO中等位基因rs9939609A与MDD呈正相关。Du等[45]分析了参与RNA修饰的5个基因(METTL3/14、WTAP、ALKBH5和FTO)内的23个SNP,结果发现ALKBH5基因与MDD相关;此外,编码m6A系统的基因内的许多SNPs与MDD的焦虑、发育迟缓和认知障碍等临床症状相关。下丘脑垂体肾上腺轴、嗜神经因子等生物系统作为基因调节器的m6A化学修饰,可以调控相关蛋白含量应答压力刺激和环境因素。基因编辑诱导并产生神经细胞,可建立起正常功能的神经元信号通路,从而有效地代表神经精神疾病的相关方面,这一策略可以为个体化疾病开辟新的途径。
4.6 癫痫 癫痫(epilepsy,EP)是由脑部神经元高度同步化异常放电所致的慢性短暂性脑功能障碍综合征[46]。由于病变部位与传导范围不同,癫痫的临床表现复杂多样,越来越多的研究将癫痫归因于具有表观遗传功能的基因变异,这些基因的产物包括表观遗传标记的编码、消除和读取,以及其他表观遗传过程等。成熟的miRNA与mRNA形成碱基对。Rowles等[47]发现miRNA-134含有一个潜在的m6A位点,在癫痫(TLE)动物模型的颞叶皮层组织以及在颞叶癫痫和海马硬化患者的海马组织中一致上调,抑制LIM激酶1(Limk1),从而抑制翻译,导致癫痫。Zheng等[48]在研究新型脯氨酰-4-羟化酶抑制剂时,发现其可以抑制FTO,提高m6A甲基化水平,并抵抗小鼠惊厥。低氧诱导因子(HIF)干预促红细胞生成素表达,降低FTO水平,促进m6A合成,发挥抗癫痫作用[49]。Tan等[50]发现丙戊酸钠治疗可增加海马区FTO的表达,经过复杂的生物过程,抑制癫痫的发生。这些结果表明m6A与癫痫之间存在关联,若通过靶向调控分子水平,保护受损区域的神经元稳态,可有效阻止癫痫的发生,这将为临床治疗癫痫提供新的思路。
4.7 肌萎缩侧索硬化(amyotrophic lateral sclerosis,ALS) ALS又称为“渐冻症”,是上下运动神经元损伤之后,四肢躯干肌肉逐渐无力或萎缩的运动神经元病[51]。ALS的发病机制目前尚不明确,可能与遗传、自身免疫力等原因有关,临床、生化和遗传特征的显著变异性也使治疗靶点的鉴定复杂化。Yoneda等[52]发现结合蛋白TLS/FUS不包含YTH特殊结构域,核定位信号(NLS)突变,迫使TLS/FUS液-液相分离(LLPS),通过LLPS形成的细胞毒性聚集物引起神经元细胞凋亡或坏死以及神经元系统的紊乱。Mcmillan等[53]不仅发现大多数结合蛋白TDP43底物携带m6A修饰,而且YTHDF2在小鼠模型和成人神经元ALS模型中促进了TDP43相关的毒性。另外,在散发性ALS患者死后脊髓组织中检测到广泛的RNA高甲基化。这些研究数据强调了m6A修饰与ALS发病机制之间的基本联系,假设通过靶向促进神经细胞分化成可代替受损神经元的新生神经元,这将为临床治疗ASL提供新的诊疗途径。
4.8 脑动静脉畸形(Brain arteriovenous malformations,AVMs) AVM是一种危及生命的先天性脑血管疾病,临床以脑出血、癫痫和中风为特征。通过对脑动静脉畸形患者的转录组学因子水平分析,Wang等[54]发现METTL3在较大的脑动静脉畸形病理组织中表达水平降低;同年研究发现WTAP及其靶基因DSP在m6A修饰脑动静脉畸形患者中表达减少,促进DSP mRNA降解,从而抑制血管生成[55]。此过程中,IGF2BP1/3蛋白可以提高DSP mRNA的稳定性。WTAP缺乏引起的WT1活性增加导致β-catenin的大量降解,这也可能抑制内皮细胞的血管生成[55]。m6A修饰可调控细胞的分化方向,控制脑血管生成,为预防脑型AVM的形成和发展提供合适的药理学靶点。
RNA甲基化修饰在调节哺乳动物发育阶段的细胞维持、分化、重新编程和控制中起着重要作用。m6A甲基化相关蛋白通过靶向调节细胞增殖周期与定向分化、转录翻译、特殊结合位点以及药物敏感等,在疾病治疗、病情延缓、预后改善等过程中发挥关键作用。这些发现为各种疾病的临床治疗开辟了新的方向。一个或多个甲基化调控因子的异常表达可能有助于开发靶向修饰蛋白的调节剂,以进一步探索在生理学和病理学中调控细胞基因表达的潜在机制。然而,在通过靶向RNA甲基化治疗疾病时,仍然有许多困难需要克服。此外,虽然RNA甲基化涉及多种模式,但对体外细胞与m6A模式结构研究有限,随着外延组学的发展,RNA中m6A修饰对于恶性疾病疗效以及预后中有望实现新的突破。
猜你喜欢
中老年保健(2021年3期)2021-12-03 02:32:25
中国民间疗法(2021年5期)2021-06-09 09:21:04
中国生殖健康(2020年7期)2020-12-10 07:48:51
饮食科学(2017年5期)2017-05-20 17:11:53
医学研究杂志(2015年7期)2015-06-22 11:01:36
现代检验医学杂志(2015年2期)2015-02-06 02:00:48
西南军医(2015年4期)2015-01-23 01:19:30
沈阳医学院学报(2014年4期)2014-12-27 13:44:30
同位素(2014年2期)2014-04-16 04:57:16
中国中医药现代远程教育(2014年20期)2014-03-01 04:31:21
