脓毒症心肌病的发病机制研究进展
2024-03-02 07:55李心瑶陈俊李灼
心血管病学进展 2024年1期
李心瑶 陈俊 李灼
(南京医科大学附属儿童医院急诊/重症医学科,江苏 南京 210008)
2016年第三次脓毒症与脓毒性休克定义的国际共识[1]将脓毒症定义为宿主对感染的反应失调而导致威胁生命的器官功能障碍。当血液循环、细胞代谢出现严重异常时,脓毒症将会进展为脓毒性休克,大幅度提高了死亡率。脓毒症心肌病(septic cardiomyopathy,SCM)是严重脓毒症和脓毒性休克常见的并发症。SCM并没有明确的定义,现阶段将SCM描述为:(1)心室扩张伴心室顺应性增加;(2)射血分数降低,心输出量没有变化;(3)对液体复苏和儿茶酚胺的反应性差;(4)心室扩张及射血分数降低可在7~10 d内恢复;(5)排除急性冠脉综合征作为病因[2]。在重症医疗领域中SCM是一个严重的临床问题,尽管近年来对SCM的认识有了很大提高,但其确切的发病机制尚未完全明了。现综述近期关于SCM发病机制的研究进展。
1 SCM的发病机制
1.1 损伤相关分子模式
心肌功能障碍是宿主对感染的反应失调,这种反应失调是由损伤相关分子模式(damage-associated molecular pattern,DAMP)和病原体相关分子模式(pathogen-associated molecular pattern,PAMP)共同驱动[3]。DAMP是内源性分子,当组织损伤后DAMP从细胞中释放出来,可作为免疫系统的有效激活剂,启动并延续非感染性炎症反应,导致全身炎症、器官损伤和死亡[4]。SCM中普遍认同的DAMP包括高迁移率族蛋白B1(high mobility group protein B1,HMGB1)、组蛋白、热休克蛋白(heat shock protein,HSP)、细胞外RNA和细胞游离DNA。
HMGB1是一种在所有哺乳动物细胞中表达的高度保守蛋白[5]。HMGB1可通过偶发性坏死或受调控的细胞死亡过程(如细胞坏死、细胞焦亡、铁死亡或细胞凋亡)导致体细胞细胞质膜破坏从而被动释放。此外,HMGB1也可通过细胞质囊泡主动释放[4]。最新研究[6-8]表明,脂多糖(lipopolysaccharide,LPS)刺激心肌细胞后HMGB1表达增加,并且抑制HMGB1活性,可缓解自噬流受阻,减少心肌细胞凋亡。细胞外组蛋白作为内源性DAMP,可能以补体(C5a)依赖性的方式出现,脓毒症通过释放C5a引起补体激活,并与其受体(C5aR1和C5aR2)相互作用,导致心肌细胞中NOD样受体热蛋白结构域相关蛋白3(NOD-like receptor pyrin domain containing 3,NLRP3)炎症小体和丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)的激活,以及细胞外组蛋白的出现。这些引起心肌细胞中心肌肌质网/内质网钙ATP酶2(sarco/endoplasmic reticulum Ca2+-ATPase 2,SERCA2)、线粒体钠钙交换蛋白(Na+/Ca2+exchanger,NCLX)和钠钾ATP酶减少,活性氧(reactive oxygen species,ROS)、细胞内Ca2+累积以及细胞因子释放,最终导致心肌细胞功能障碍[9]。HSP是维持细胞内稳态的分子伴侣,对免疫反应具有强大的作用[10]。有研究[11]表明,HSP70通过改善线粒体功能障碍和抑制NLRP3炎症小体介导的细胞焦亡来改善脓毒症诱导的心肌功能障碍。同时,HSP70也可抑制自噬激活来减少心肌细胞凋亡[12]。除此之外,细胞外RNA及细胞游离DNA释放到细胞外后可作为DAMP促进炎症发展。
1.2 PAMP
LPS也称为内毒素,是革兰氏阴性菌外膜的主要成分。LPS在细菌细胞壁破坏后被释放出来,并以细菌外膜囊泡的形式活跃分泌。LPS信号通路主要涉及LPS与特异性免疫细胞受体的相互作用。Toll样受体(Toll-like receptor,TLR)家族用于识别细菌PAMP,以及细胞质部分用于细胞内信号传导。LPS结合TLR4和髓样分化蛋白2,即TLR4-髓样分化蛋白2复合物,衔接蛋白随后被招募到复合物的胞内区域,激活髓样分化因子依赖性和非依赖性通路。白细胞介素(interleukin,IL)-1受体衔接蛋白和髓样分化蛋白88衔接,激活核因子κB (nuclear factor-κB,NF-κB) 和MAPK,导致促炎细胞因子的产生。严重的细菌感染和血清中LPS的突然增加,使炎症因子不受控制地释放和免疫反应不适当地扩增,导致组织损伤、器官衰竭和死亡[13-14]。
1.3 线粒体功能障碍
心脏需大量的ATP来维持正常的收缩和舒张功能,95%的ATP来自于线粒体的氧化磷酸化[15-16],因此线粒体功能障碍导致的能量代谢衰竭可能参与SCM的发生发展(图1)。
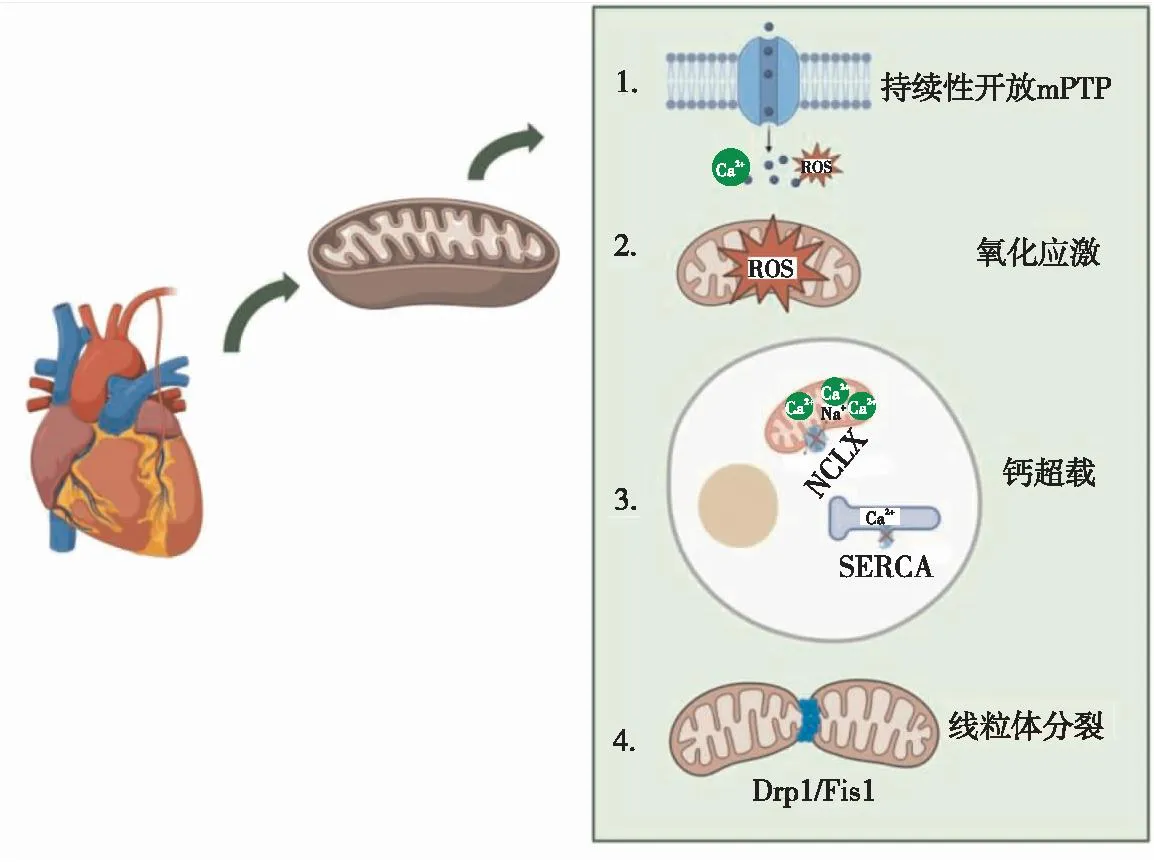
注:mPTP,线粒体膜通透性转换孔;Drp1,线粒体动力相关蛋白1;Fis1,线粒体分裂蛋白1。图1 线粒体功能障碍参与SCM的发病机制
1.3.1 线粒体超微结构的改变
线粒体膜通透性转换孔是一种高电导通道,暂时开放(可逆)可使Ca2+及ROS转运到细胞质中,持续性开放(不可逆)使线粒体内膜对离子和小分子溶质的渗透性突然增加,并允许代谢物质进入线粒体基质,这导致氧化磷酸化的解偶联,ROS增加,ATP的生产效率降低[17-19]。最新研究[20-21]表明,通过抑制线粒体膜通透性转换孔的开放和限制心脏组织钙含量超载,可改善心肌结构和功能障碍。
1.3.2 氧化应激
ROS是正常生理过程产生的产物,线粒体呼吸链复合体Ⅰ和Ⅲ生理性产生少量ROS,生理浓度下ROS在细胞信号传导和维持组织稳态中发挥重要作用[22-23]。单个线粒体产生的ROS会诱导其他线粒体产生更多的ROS,这种现象被称为ROS诱导的ROS释放现象,过量的ROS产生被称为氧化应激[24]。脓毒症可通过增加氧化应激和过量产生线粒体ROS(mtROS),导致线粒体功能障碍。过量的ROS会损伤细胞,并参与各种器官功能障碍。研究[22]表明,ROS会导致SCM中氧化磷酸化通路的特异性损伤(如线粒体呼吸链复合体Ⅰ、复合体Ⅳ和F0F1功能障碍),尤其是心肌细胞线粒体损伤。ROS可诱导NLRP3从细胞核向细胞质转运,解离性硫氧还蛋白互作蛋白可直接与细胞质NLRP3相互作用,形成炎症小体,引起心肌细胞损伤[25]。抑制ROS累积可阻断NLRP3介导的心肌细胞凋亡和焦亡,改善心肌功能。目前研究[26]发现,药物通过抑制mtROS的产生可改善LPS诱导的心肌功能障碍,为干预SCM提供了新的治疗靶点。
1.3.3 钙超载
在脓毒症的早期阶段,大量的Ca2+从心肌细胞外流入细胞质,导致细胞质中钙超载,最终诱导线粒体钙超载。钙超载引起的线粒体损伤是脓毒症心肌功能障碍的重要机制。NCLX介导的线粒体Ca2+外排受阻是线粒体钙超载的重要因素[27]。最近研究[28]验证了脓毒症期间PINK1-PKA-NCLX轴在心肌细胞线粒体钙超载中起重要作用。另外,SERCA2a负责Ca2+从细胞质转运到肌质网来维持低的胞质钙水平,脓毒症中SERCA活性降低而导致胞质钙水平升高,进而导致线粒体钙超载[29-30]。有研究[31]表明,心肌细胞丙酮酸激酶M2直接与SERCA2a相互作用并调节其蛋白表达。丙酮酸激酶M2缺乏导致SERCA2a下调后钙失衡、心肌细胞凋亡和心肌收缩障碍。通过限制钙超载可减轻脓毒症中心脏结构改变和功能障碍[32]。
1.3.4 线粒体动力学机制
线粒体动力学是指线粒体结构的动态变化,包括线粒体形态、线粒体在细胞内的分布以及它们沿着细胞骨架的运动。线粒体不断分裂和融合,形成一个动态的线粒体网络,适应代谢变化,保持细胞完整性,防止自噬[33]。在细胞中,线粒体分裂和融合之间存在高度调节的平衡,从而维持正常的线粒体功能。线粒体分裂是由线粒体动力相关蛋白1(dynamin-related protein 1,Drp1)和线粒体分裂蛋白1调控[34]。在生理条件下,Drp1主要存在于细胞质中,但在激活后,Drp1被招募到线粒体表面寡聚化与相应受体结合,诱导线粒体分裂。尽管生理水平的分裂对于线粒体质量控制是必要的,但过度分裂会导致线粒体功能障碍和氧化应激增加。抑制Drp1和线粒体分裂蛋白1的相互作用可减少病理性线粒体分裂,促进ATP生成,并预防心肌功能障碍,提高脓毒症小鼠的存活率[35]。Drp1介导的线粒体分裂可促进SCM的发生发展[36],同时,抑制Drp1活性可改善线粒体功能,从而减轻心肌抑制和心肌细胞死亡[37-38]。
线粒体融合由线粒体融合蛋白1/2和视神经萎缩相关蛋白1介导。目前尚无线粒体融合相关蛋白参与SCM发病机制的研究。
1.4 铁死亡
铁死亡是近年来发现的一种特殊细胞死亡形式,涉及正常生理及多种病理过程。形态学方面,铁死亡表现为线粒体体积减小、内外膜密度增加和线粒体嵴减少或消失,但细胞膜保持完整,细胞核大小正常,染色质无浓度变化;生物化学方面,细胞内谷胱甘肽(glutathione,GSH)耗竭,谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)活性降低,脂质过氧化物不能被GPX4催化的还原反应代谢,Fe2+通过芬顿反应氧化脂质,产生大量ROS,促进铁死亡。胱氨酸/谷氨酸反转运体是一种氨基酸反转运蛋白,是由两个亚基SLC7A11和SLC3A2组成的异源二聚体,参与GSH的合成[39]。
目前研究[40]表明,LPS增加了细胞核受体共激活因子4(nuclear receptor coactivator 4 ,NCOA4)的表达,NCOA4可直接与铁蛋白相互作用,并以噬铁蛋白依赖的方式降解铁蛋白,从而释放出大量的游离铁,进而导致mtROS的产生和铁死亡。抑制铁死亡可减轻线粒体损伤,提高脓毒症小鼠的心脏功能和存活率。脓毒症中GSH活性、GPX4活性以及SLC7A11蛋白表达水平显著降低,导致铁死亡诱导的氧化应激和脂质过氧化。而硫氢化钠可通过提高GSH水平减轻心肌氧化应激和脂质过氧化来保护脓毒性心肌损伤[41]。白藜芦醇通过上调Sirt1/Nrf2途径减少SCM铁死亡来改善心肌功能障碍[42]。LPS增加了p53和铁蛋白的表达,但降低了GPX4和SLC7A11的水平。跨膜蛋白43与之作用相反,并且通过抑制铁死亡来防止SCM[43]。铁死亡抑制剂1是铁死亡的特异性抑制剂,通过抑制TLR4/NF-κB信号通路改善心肌功能障碍[44]。环维黄杨星D(cyclovirobuxine D,CVB-D)可通过抑制氧化应激、铁过载、脂质过氧化和Ca2+内流来减少脓毒症诱导的心肌细胞铁死亡。在机制上,CVB-D通过IL-6/STAT3/铁调素轴促进铁转运蛋白介导的细胞内铁释放来抑制铁死亡。此外,CVB-D被证明通过激活心肌细胞中的Nrf2/SLC7A11/GPX4轴来抑制铁死亡[45]。
1.5 细胞焦亡
在细胞和分子水平上,细胞焦亡是SCM的重要病理生理学机制。细胞焦亡的经典形式是由炎症小体激活的胱天蛋白酶(caspase)家族和Gasdermin蛋白家族Gasdermin D蛋白介导。NLRP3是启动免疫反应和炎症小体形成的重要介质,促进促炎细胞因子(如caspase-1、IL-1β以及IL-18)的成熟和分泌,并引发焦亡。在脓毒症中,焦亡可导致细胞膜破裂,释放多种炎症因子。研究发现,TLR4激活STING-IRF3途径,通过敲低STING有效抑制LPS诱导后心肌中NLRP3的表达,而与心肌细胞焦亡相关的caspase-1、IL-1β和IL-18等炎症因子的蛋白水平也降低。然而,LPS诱导的ROS释放促进NLRP3向细胞质转移,并导致硫氧还蛋白互作蛋白与炎症小体结合,启动焦亡途径,导致心肌细胞死亡。总体而言,细胞焦亡参与SCM的发生发展,通过靶向焦亡机制或上游靶向因子调节剂以直接或间接的方法抑制细胞焦亡会对心肌细胞产生保护作用[25,46-47]。
2 总结
目前SCM的发病机制暂不明确,但可见SCM是由多方面因素导致的疾病,微生物毒素通过多种途径诱导心肌损伤,线粒体功能障碍在SCM的发生发展中起重要作用,通过氧化应激、Ca2+失衡等方面加重心肌细胞损伤。除此之外,铁死亡作为近年来新发现的细胞死亡形式受到广泛关注,其中的机制尚需更多的深入研究阐述。这些机制的阐明有助于发现SCM新的干预靶点,改善脓毒症患者的预后。
猜你喜欢
科学导报(2024年20期)2024-04-22
世界科学技术-中医药现代化(2022年3期)2022-08-22
医学综述(2022年7期)2022-04-19
昆明医科大学学报(2020年12期)2021-01-26
中华养生保健(2020年4期)2020-11-16
世界科学技术-中医药现代化(2020年2期)2020-07-25
中国中医急症(2019年10期)2019-05-21
中华老年多器官疾病杂志(2016年9期)2016-04-28
上海农业学报(2016年5期)2016-02-10
中国蔬菜(2015年9期)2015-12-21