低氧诱导因子-1在低氧性肺动脉高压中的研究进展
2024-03-02 07:55金鸿锦卢义丁彦春
心血管病学进展 2024年1期
金鸿锦 卢义 丁彦春
(大连医科大学附属第二医院,辽宁 大连 116021)
低氧性肺动脉高压(hypoxic pulmonary hypertension,HPH)是由缺氧引起的肺动脉压力进行性升高的肺血管疾病。长期慢性HPH可引起右心室后负荷增加,最终导致不可逆性右心衰竭甚至死亡,严重威胁患者生命。肺血管受到低氧刺激时的早期反应为肺血管收缩,称为低氧性肺血管收缩(hypoxic pulmonary vasoconstriction,HPV),其生理意义在于减少缺氧肺组织的血流,将血流重新分配至通气较好的肺组织,从而维持正常的通气/血流比值,保持良好的气体交换功能[1]。然而,长期暴露于低氧环境可引起机体慢性缺氧,慢性缺氧通过诱导肺动脉平滑肌细胞(pulmonary artery smooth muscle cells,PASMC)和肺动脉内皮细胞(pulmonary artery endothelial cells,PAEC)增殖、凋亡失衡和代谢改变,引起不可逆性的低氧性肺血管重塑(hypoxic pulmonary vascular remodeling,HPVR),最终导致肺动脉高压的形成和进展。因此,HPV和HPVR是HPH发生和发展过程中的关键病理生理机制。
低氧诱导因子(hypoxia inducible factor,HIF)-1是维持细胞氧稳态的核心转录因子,可调控数百个氧依赖性靶基因的转录。HIF-1可促进细胞糖代谢模式的转变,调节细胞膜表面离子通道活性、肺血管收缩及舒张因子活性等,调节机体对缺氧的适应性反应,参与HPH的发生和进展。现对HIF-1及其下游信号分子在HPH发生和发展中的作用机制进行综述。
1 低氧状态下HIF-1产生增多的机制
HIF-1是高度保守的转录因子,几乎表达于所有的组织细胞中。HIF-1是由具有氧感知功能的HIF-1α亚基和组成性表达的HIF-1β亚基构成的异源性二聚体。HIF-1α是HIF-1的活性亚基,其表达水平受细胞内氧浓度的调节。HIF-1α中含有两种不同的羟基化位点,分别是2个位于氧依赖性降解结构域的脯氨酸残基(Pro402和Pro564)和1个位于C端转录激活域的天冬酰胺残基(Asn803),可分别被脯氨酸羟化酶(prolyl hydroxylase,PHD)及HIF抑制因子羟基化[2]。常氧状态下,PHD能诱导HIF-1α脯氨酸残基羟基化[3],然后与von Hippel-Lindau蛋白结合,招募E3泛素蛋白连接酶使其泛素化,最后通过蛋白酶体降解[4],导致HIF-1α的表达减少。HIF抑制因子是调节HIF-1α转录活性的关键蛋白,可促进HIF-1α天冬酰胺残基羟基化,阻止HIF-1与缺氧反应元件中辅助因子p300/CREB结合蛋白结合,从而抑制HIF下游靶基因的转录[5]。故常氧状态下,HIF-1的表达水平及转录活性均较低。
低氧状态下,PHD的活性降低,相关机制可能包括:(1)PHD诱导HIF-1α羟基化的过程需要氧气参与,因此低氧状态下PHD引起HIF-1α的羟基化减少。(2)低氧状态下,三羧酸循环中琥珀酸脱氢酶活性降低,导致琥珀酸转换为延胡索酸减少,线粒体中琥珀酸盐水平升高,被琥珀酸转运蛋白转运至细胞质内[6],琥珀酸盐对PHD具有浓度依赖性的抑制作用[7]。(3)低氧可能诱导线粒体复合体Ⅲ产生活性氧(reactive oxygen species,ROS)[8],大量的ROS从线粒体转运至细胞质内,ROS可能导致PHD3的活性降低[9]。故低氧状态下PHD活性降低,引起HIF-1α羟基化减少,导致HIF-1α降解减少、稳定性增加,HIF-1α进入细胞核内与HIF-1β结合形成稳定的HIF-1二聚体,HIF-1二聚体再与靶基因的缺氧反应元件结合,通过p300/CREB结合蛋白辅助因子的衔接,形成转录复合物,与低氧反应元件结合,激活下游靶基因的转录过程[10],参与HPH的发生和进展。
2 HIF-1参与HPH的机制
2.1 HIF-1引起细胞糖代谢模式的转变
在HPH中观察到,PASMC及PAEC表现出与肿瘤细胞相似的糖代谢模式的转变,即使在有氧条件下也会表现为糖酵解途径增强而糖有氧氧化减弱,该现象称为Warburg效应[11]。HIF-1可调控多种与糖酵解过程相关的酶的活性,促进细胞糖代谢模式由糖有氧氧化转变为糖酵解,其机制包括:(1)HIF-1上调葡萄糖转运蛋白的表达,促进细胞对葡萄糖的摄取,但由于糖有氧氧化减少,细胞内葡萄糖含量的增加为糖酵解途径提供了更多的底物,促进了糖酵解途径[12]。(2)HIF-1促进糖酵解关键酶包括己糖激酶、磷酸果糖激酶-1、丙酮酸脱氢酶激酶(pyruvate dehydrogenase kinase,PDK)-1的表达,促进糖酵解过程。PDK-1可通过磷酸化抑制丙酮酸脱氢酶的活性,使得丙酮酸分解为乙酰辅酶A的数量减少,进入三羧酸循环的乙酰辅酶A不足,抑制了糖有氧氧化过程,并将丙酮酸重新导向糖酵解过程[5]。此外,HIF-1可促进6-磷酸果糖-2-激酶/果糖-2,6-二磷酸酶3(6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3,PFKFB3)基因的表达,PFKFB3 可促进果糖-6-磷酸转化为果糖-2,6-二磷酸,而果糖-2,6-二磷酸是磷酸果糖激酶-1最有效的变构激活剂[13],PFKFB3介导的糖酵解过程促进PAEC释放生长因子和促炎细胞因子,促进PASMC的增殖和肺血管的炎症反应[14]。(3)HIF-1促进乳酸脱氢酶的表达,加速丙酮酸分解为乳酸,促进细胞的糖酵解过程。HIF-1对于糖酵解过程中关键酶的调节如图1。
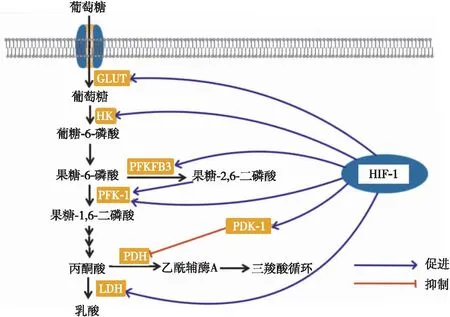
注:GLUT,葡萄糖转运蛋白;HK,己糖激酶;PFK-1,磷酸果糖激酶-1;PDH,丙酮酸脱氢酶;LDH,乳酸脱氢酶。图1 HIF-1调控糖酵解过程相关酶的示意图
HIF-1参与Warburg效应的机制可能与磷脂酰肌醇3激酶/蛋白激酶B/哺乳动物雷帕霉素靶蛋白(phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin,PI3K/Akt/mTOR)信号通路有关。PI3K/Akt/mTOR信号通路在调节Warburg效应中发挥关键作用,mTOR通过促进HIF-1的表达放大Warburg效应[15],血小板源性生长因子可通过激活PASMC增殖中的PI3K/Akt/mTOR/HIF-1α通路促进Warburg效应[16]。HIF-1可能作为PI3K/Akt/mTOR通路的下游信号分子,调节糖酵解过程中关键酶的活性,促进细胞摄取葡萄糖,提高糖酵解过程中关键酶的活性,从而促进葡萄糖的糖酵解过程。且HIF-1可使丙酮酸分解为乙酰辅酶A的数量减少,减少了三羧酸循环的原料,抑制了糖有氧氧化的过程。因此,HIF-1的激活是促进细胞糖代谢模式转变的重要开关。糖有氧氧化途径受到抑制能减少缺氧引起的ROS产生,降低了ROS对于细胞内蛋白质、脂质和DNA等大分子的毒性作用[17]。糖酵解途径的增强导致PASMC能量产生效率降低,能量供应相对不足,使PASMC表现出过度增殖及抗凋亡的特性[18],糖酵解途径导致乳酸堆积,通过上调组蛋白乳酸化而促进PASMC的增殖[9]。HIF促进细胞糖代谢模式的转变,其意义在于减少细胞耗氧量,适应HPH引起的低氧环境,但也同时引起PASMC的增殖和凋亡失衡,导致了肺血管重塑的发生。
2.2 HIF-1调节细胞膜表面离子通道活性
细胞内钙离子浓度增加是引起PASMC收缩的主要因素,也是PASMC增殖的重要刺激因子。细胞内钙离子浓度的变化主要由细胞膜表面不同类型的钙通道介导,包括电压依赖性钙通道(voltage-dependent calcium channels,VDCC)、钙库操纵性钙通道及受体门控性钙通道等。HIF-1可调节PASMC细胞膜表面钙通道及钾通道的活性,引起PASMC细胞膜内外钙离子和钾离子浓度的改变,导致PASMC收缩和异常增殖,进而导致HPV和HPVR的发生。
细胞膜去极化是引起VDCC开放的重要因素,电压门控钾通道(voltage-gated potassium channel,KV)的表达变化可导致PASMC膜电位的改变。Whitman等[19]研究发现低氧状态下HIF-1通过促进内皮素(endothelin,ET)-1的表达,导致PASMC中KV1.5、KV2.1的表达减少及细胞内钙离子浓度的增加,通过ET-1受体抑制剂BQ123治疗可削弱上述效应。芹菜素通过抑制HIF-1α-KV1.5途径,诱导PASMC线粒体依赖性凋亡,从而减轻缺氧诱导的肺动脉高压的进展[20]。因此,HIF-1可能通过抑制KV的开放,导致钾离子外流减少,细胞内钾离子浓度增加,PASMC膜去极化,进一步引起VDCC开放、细胞外钙离子内流、细胞内钙离子浓度增加,从而导致肺血管收缩。
钙库操纵性钙通道是由于内质网和肌质网中钙离子浓度降低而被激活的细胞膜表面的钙通道。瞬时受体电位通道(transient receptor potential channel,TRPC)是钙库操纵性钙通道的重要组成部分,其中包括TRPC1~7 七个成员[21]。通过抑制TRPC1、TRPC3、TRPC4和TRPC6能抑制PASMC的增殖,抑制TRPC1可减少PASMC的迁移能力,抑制TRPC1、TRPC3、TRPC4和TRPC6可促进细胞凋亡,表明TRPC参与了肺动脉高压过程中PASMC的增殖、迁移及凋亡抵抗过程[22]。BI-749327是一种具有高选择性的TRPC6拮抗剂,可抑制PASMC中PI3K/Akt/mTOR信号通路,阻断TRPC6介导的钙离子内流,从而抑制PASMC的增殖,进而降低肺动脉压力并部分逆转肺血管重塑[23]。HIF-1在调节PASMC中TRPC表达和钙离子稳态方面具有重要作用,HIF-1可促进TRPC的表达,引起细胞外钙离子内流,导致细胞内钙离子浓度增加。低氧状态下,HIF-1可能通过诱导PASMC中骨形态发生蛋白4的表达,通过ERK1/2和p38MAPK信号通路诱导TRPC1、TRPC6的表达增加及细胞内钙离子浓度增加,参与HPH的发生[24]。
2.3 HIF-1调节肺血管收缩及舒张因子活性
肺血管收缩及舒张因子的平衡失调是肺动脉高压的特征之一。HPH中HIF-1可调节PAEC分泌的血管收缩及舒张因子的活性,导致血管收缩因子包括ET、5-羟色胺、血管紧张素、血栓素等增加,而血管舒张因子包括一氧化氮(nitric oxide,NO)、前列环素和血管活性肠多肽减少,并通过旁分泌的方式作用于PASMC[25-26]。其中,ET-1和NO二者之间的平衡失调在HPH的发生过程中具有重要作用。HIF-1通过在转录水平上诱导下游靶基因ET-1及诱导型一氧化氮合成酶(inducible nitric oxide synthase,iNOS)的表达,影响了ET-1与NO的平衡,从而导致持续的肺血管收缩。
ET-1通过与ET-1受体结合发挥作用,包括ET-A受体及ET-B受体。ET-A受体主要表达于PASMC,介导PASMC的增殖和血管收缩,而ET-B受体主要位于PAEC,通过释放NO等血管舒张因子导致血管舒张。ET主要由PAEC产生,通过旁分泌的途径作用于PASMC[27]。ET-1基因的启动子区域有HIF-1α的特异性结合位点,低氧可能通过促进HIF-1α产生增多而诱导ET-1基因的表达,使ET-1分泌增加。ET-1亦可通过增加细胞内钙内流、促进ROS的产生及激活ERK1/2通路等机制,上调大鼠PASMC中HIF-1的合成并下调PHD2介导的HIF-1α的降解,从而促进PASMC中HIF-1的表达[28]。因此,HIF-1和ET-1形成一个双向调节回路,在肺血管重塑中起重要作用[29]。
NO是PAEC合成及释放的具有强扩血管作用的内皮源性血管活性因子,iNOS是NO合成过程中的限速酶。低氧状态下HIF-1表达增多,进而与iNOS基因启动子区域的相应位点结合,诱导iNOS mRNA的表达,促进NO的合成。当ET-1与PAEC上的ET-B受体结合后,亦可促进PAEC释放NO,发挥血管舒张作用。然而,当机体长期处于慢性缺氧状态时,PAEC分泌NO减少,其可能机制包括:(1)大量的ET-1可抑制 HIF-1与iNOS靶基因的特异性位点结合,抑制HIF-1诱导的iNOS mRNA表达,导致PAEC合成NO减少[30]。(2)慢性缺氧可产生大量的ROS,直接损伤PAEC,导致PAEC合成NO减少。以上机制均可导致ET-1与NO之间的平衡失调,引起肺血管收缩,进而导致HPH的发生。
3 针对HIF-1治疗HPH的靶点
目前已有多项研究发现,针对HIF-1及其下游信号分子途径的药物能降低肺动脉压力、延缓肺血管及右心室重塑。二氯乙酸是PDK抑制剂,能促进丙酮酸转化为乙酰辅酶A,抑制糖酵解进而促进糖有氧氧化过程。Li等[31]发现,使用二氯乙酸处理人PASMC可减少HIF-1α的表达,并抑制PDK-1和己糖激酶-2的活性,从而逆转Warburg效应,导致PASMC的增殖减少和凋亡增加。法舒地尔是选择性Rho激酶抑制剂,通过法舒地尔抑制PASMC中HIF-1α的表达可显著抑制缺氧诱导的TRPC1和TRPC6的表达[32],且法舒地尔可抑制HPH大鼠的HIF-1α及ET-1表达,促进NO的表达,延缓右心室重塑,抑制HPH的进展[33]。拓扑替康为拓扑异构酶Ⅰ抑制剂,能抑制HIF-1的转录,拓扑替康通过抑制缺氧诱导的大鼠PASMC中HIF-1α、TRPC1、TRPC4、TRPC6的表达和钙离子内流,从而抑制PASMC的增殖、迁移和收缩合成表型转换,减轻肺血管重塑和右心室肥厚[34]。Dai等[35]首次发现了针对ET-A受体的免疫疗法(包括ETRQβ-002疫苗和针对ETR-002的特异性抗体),该疗法可显著使HPH小鼠右心室收缩压降低10 mm Hg(1 mm Hg=0.133 3 kPa),有效抑制HPH小鼠的肺小动脉及右心室重塑,降低右心室收缩压,且不引起肝肾功能损害。
4 结语和展望
HPH是一种严重的肺血管疾病。目前临床上针对HPH的药物治疗主要是通过增强NO和前列环素途径的信号转导或减弱ET途径的信号转导,恢复血管收缩因子和血管舒张因子的平衡[36],达到扩张肺血管、降低肺动脉压的目的。虽然扩血管药物能改善HPH患者症状,但不能抑制或延缓肺血管重塑,而肺血管重塑是HPH发展且难以治愈的主要原因,故目前尚不能通过药物治疗治愈HPH。HIF-1是维持细胞氧稳态的关键转录因子,本文对于HIF-1及其下游信号分子途径在HPH发生和发展过程中的作用进行综述,表明HIF-1可通过促进细胞糖代谢模式的转变,调节细胞膜表面离子通道活性、血管收缩及舒张因子活性等,参与HPH的发生和进展,其机制可能涉及PI3K/Akt/mTOR信号通路的异常激活,但其具体机制仍有待于进一步探究,仍需更多的研究验证HIF-1及其下游信号分子途径在HPH患者中的作用机制及治疗效果。
猜你喜欢
中南医学科学杂志(2022年6期)2022-12-04
天津医科大学学报(2021年3期)2021-07-21
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
中国循证心血管医学杂志(2020年11期)2020-01-08
中华老年口腔医学杂志(2016年4期)2017-01-15
中国医学装备(2016年6期)2016-12-01
国外医药(抗生素分册)(2016年5期)2016-07-12
中国医药生物技术(2015年4期)2015-12-26
实验动物与比较医学(2014年5期)2014-02-28