
SPE-HPLC-UV 法测定绞股蓝提取物中皂苷的含量
2024-02-05 08:04杨悠悠赵青余张军民余雅男
中国饲料 2024年3期
陈 晗, 杨悠悠, 赵青余, 张军民, 余雅男 , 张 凯
(1.中国农业科学院北京畜牧兽医研究所,动物营养学国家重点实验室,北京100193;2.青岛农业大学食品科学与工程学院,山东 青岛 266109;3. 青岛特种食品研究院,山东 青岛 266109)
绞股蓝(Gynostemma pentaphyllum)是葫芦科绞股蓝属多年生草质藤本药食两用植物,《中华本草》记载,绞股蓝别名为七叶胆、小苦药、公罗锅底、落地生等(国家中医药管理局《中华本草》编委会,1999),在日本还被称为“甘茶蔓”。我国绞股蓝的三大产区分别为陕西安康平利、 广西金秀和湖北神农架地区, 分布广泛, 资源丰富 (李华等,2015)。 绞股蓝在我国具有悠久的药用历史,根据《本草纲目》记载,其具有消炎解毒、止咳祛痰、清热补虚的功效。 在1986 年的国家科委的“星火计划” 中, 由于突出的药用价值绞股蓝位居待开发“名贵中药材”之首(史琳等,2011),并在2020 年被卫生部纳入保健品名录。 绞股蓝含有多种生物活性成分,包括皂苷、黄酮类化合物、多糖、氨基酸、微量元素以及其他多种化学成分(鲍凤霞等,2018),绞股蓝皂苷一直是研究热点,并且被认为是绞股蓝中主要发挥生物活性的物质。 研究表明绞股蓝含有Rb1、Rb3、Rc、Rd、F2、Rg3 等多种皂苷, 具有相似的骨架结构 (Su 等,2021;Ha 等,2019),都属于达玛烷型四环三萜,且部分绞股蓝皂苷在体内可以转化为稀有的人参皂苷(郑溢等,2018)。 因此绞股蓝皂苷,具有神经保护(Park 等,2020)、抗氧化(Seo 等,2017)、抗癌症(Ma 等,2019;Xing 等,2018)、抑菌(Srichana 等,2011)、抗衰老(Phu 等,2020)、降血糖(Zhu 等,2021)、调节血脂、 保护肝脏、 调节肠道微生物 (Zhang 等,2021;Huang 等,2019;Khan 等,2019;Bae 等,2018)等多种功能。在“禁抗、限抗”的大背景下,绞股蓝已列入我国《饲料原料目录》,其产品的市场需求也日益强烈。
目前,有采用UPLC-Q-Trap-MS 法对绞股蓝药材中的9 种皂苷成分含量进行测定 (Duan 等,2021),HPLC-UV-ELSD 法对绞股蓝药材中槲皮素、芦丁、绞股蓝皂苷A 和绞股蓝皂苷XLIX 4 种指标性成分的含量进行测定与分析(王源源和丁鹏,2021),RP-HPLC-ELSD 法对绞股蓝药材中的皂苷含量进行测定(李浩飞,2015),HPLC-QAMS法对药品清肺散结丸中绞股蓝皂苷ⅩLⅨ、 绞股蓝皂苷A 和绞股蓝皂苷ⅩⅦ的含量进行测定(周颖等,2020),HPLC 法对药品绞股蓝总苷分散片中绞股蓝皂苷A 的含量进行测定(徐文峰等,2019)。对绞股蓝提取物中绞股蓝皂苷含量测定的研究较少,且我国尚未制定有关饲料原料绞股蓝粗提物质量评价相关标准。本研究通过对色谱条件及前处理进行研究、优化,旨在建立SPE-HPLC-UV 法测定绞股蓝三萜皂苷含量,为绞股蓝粗提物作为饲料原料使用时,其中三萜皂苷(绞股蓝皂苷A 和绞股蓝皂苷XLIX)含量的测定提供参考。
1 材料与方法
1.1 仪器与试剂 Agilent 高效液相色谱仪(1290 Infinity II);KQ-500DE 型数控超声仪 (昆山市超声仪器有限公司);HD-2500 型多管涡旋混合仪(杭州佑宁仪器有限公司);XP-205 型分析天平(瑞士METTLER TOLEDO 公司);Avvanti J-26S XP 高速冷冻离心机;SPE 固相萃取萃取柱:Cleanert S C18(60 mg/ 3 mL 50/pkg);固液萃取减压净化系统。
乙腈和甲醇均为色谱纯,均购于Fisher 试剂公司;正丁醇和乙醇为色谱纯,均购于Macklin 试剂公司;屈臣氏蒸馏水;绞股蓝XLIX 标准对照品和绞股蓝A 标准对照品来自军事医学科学院马百平研究员团队(纯度≥98.0%)。市场采购绞股蓝提取物样品6 批,编号为S1 ~S6,规格均为提取率20∶1。
1.2 色谱条件 色谱柱:Kinetex C18色谱柱(150 mm× 4.6 mm,2.6 μm);柱温:40 ℃;以水-乙腈为流动相;流速:1.0 mL/min;进样量:10 μL;梯度洗脱见表1;检测器:紫外检测器;检测波长:203 nm。混合对照品高效液相色谱图见图1。
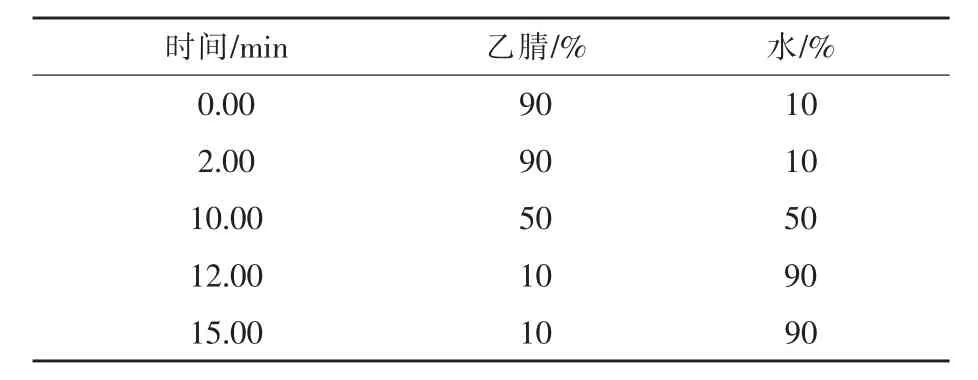
表1 流动相梯度洗脱
1.3 溶液配制
1.3.1 标准品溶液配制 精密称取并记录绞股蓝皂苷XLIX 和皂苷A 标准对照品, 用色谱纯甲醇定容至1 mL,摇匀,得对照品贮备液。 储存于-20 ℃冰箱备用, 临用时再配制成为不同浓度梯度的混合工作液。
1.3.2 供试品溶液制备 采用甲醇直接提取法对绞股蓝提取物样品进行制备。 精密称取样品约0.2 g,置于50 mL 离心管中,加10 mL 75%甲醇水,2500 r/min 振摇30 min,50 kHz 超声30 min,8000 r/min、4 ℃离心5 min,过0.45 μm 有机相膜后,取上清1 mL 用蒸馏水定容至10 mL,作为绞股蓝提取液。
1.3.3 供试品溶液净化 将SPE 柱置于固相萃取系统上,活化:向SPE 柱中加入3 mL 甲醇,浸润后缓慢过柱弃流出液,再加入3 mL 蒸馏水,浸润后缓慢过柱弃流出液;上样:精密吸取0.5 mL待净化绞股蓝提取液至已活化的SPE 柱中,以2 ~3 滴/s 流速过柱,弃流出液;淋洗:依次加入2 mL蒸馏水,2 mL 20%乙腈,淋洗SPE 柱,弃淋洗液;洗脱:加入2 mL 40%乙腈,浸润后以2 ~3 滴/s流速过柱,收集洗脱液,待液相色谱测定。
2 结果与讨论
2.1 色谱条件优化 比较Shimadzu Sum C18液相色谱柱(250 mm×4.6 mm,5 μm)和Kinetex C18液相色谱柱(150 mm×4.6 mm,5 μm)2 款不同色谱柱,发现在相同的条件下,2 种色谱柱都具有较好的分离度,但Kinetex C18液相色谱柱(150 mm×4.6 mm,5 μm)分离条件较好,色谱图基线平稳,色谱峰峰形对称,出峰时间也相对缩短,节省实验周期。确定色谱柱后, 考察了30、35 ℃和40 ℃不同色谱柱柱温,发现当柱温为40 ℃时,目标化合物色谱峰与相邻色谱峰分离效果更好,能提前出峰时间。
2.2 前处理条件优化 本研究考察了甲醇、乙醇和水作为直接提取法的溶剂对绞股蓝皂苷含量测定的影响,结果表明甲醇的提取效果更好,这也与龙凌云(2018)等对绞股蓝皂苷前处理溶剂一致,并对甲醇水浓度(25%、50%、75%和100%)进行选择, 比较得75%甲醇水浓度具有最高提取率,结果如图2 所示。绞股蓝提取液中成分相对复杂,含有多种皂苷、黄酮、多糖等成分,在本实验选择了固相萃取柱对目标皂苷进行净化和富集。 首先以20 μg/mL 混合对照品为模型探究SPE 柱的淋洗和洗脱条件, 将SPE 柱先后使用3 mL 甲醇和蒸馏水活化,加入2 mL 混合对照品,以蒸馏水及20%、40%、60%、80%、100%乙腈水淋洗SPE 柱,收集洗脱液,进行液相检测。 结果表明,40%乙腈条件下,目标化合物洗脱完全,对照品经SPE 柱洗脱后,绞股蓝皂苷XLIX 的回收率达91.3%,绞股蓝皂苷A 的回收率达94.8%,结果证明SPE 柱对样品前处理的的净化效果良好,且回收率较高,这也与Chen 等 (2021) 对保健品中人参皂苷和Song 等(2020)对原人参二醇人参皂苷的试验结果相似。SPE 柱有一定柱容量,过载会出现柱穿透现象,即部分目标化合物也会随流动相流出(彭晓俊等,2013),实验确定了此SPE 柱下最适添加绞股蓝提取液体积为0.5 mL。
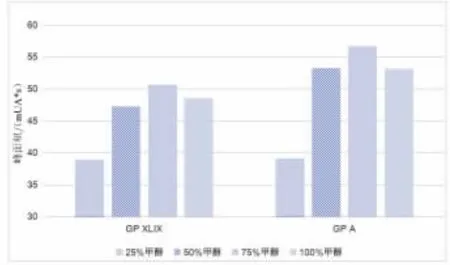
图2 不同比例甲醇水提取率比较
2.3 线性实验 用甲醇将溶剂配制成200 μg/mL混合标准使用液, 并逐级稀释至浓度梯度分别为0.1、0.5、1.0、2.5、10、25、50、100、200 μg/mL 混合对照品溶液,按“2.1”项下色谱条件进样测定,记录峰面积。 以对照品溶液的浓度为横坐标(X),峰面积为纵坐标(Y),绘制标准曲线。检出限为S/N=3 的值, 定量限为S/N=10 的值。 实验结果如表2所示, 绞股蓝皂苷XLIX 和绞股蓝皂苷A 在一定浓度范围内与峰面积的线性关系良好。
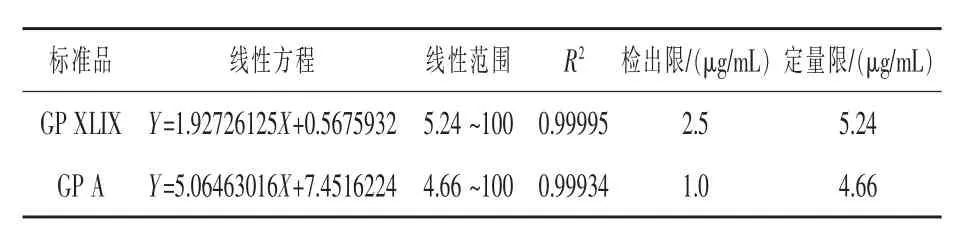
表2 线性实验结果
2.4 精密度实验 按照“2.2.1” 项方法制备100 μg/mL 混合标准贮备溶液,按“2.1”项下色谱条件进样测定, 连续6 次进样, 参照峰为绞股蓝皂苷XLIX 和皂苷A 峰, 分别计算2 个峰的相对保留时间RSD 和相对峰面积RSD。 结果显示,两个峰相对保留时间的RSD 为0.01%和0.01%, 相对峰面积的RSD 为0.32%和0.31%, 表明高效液相色谱仪的精密度良好。
2.5 稳定性实验 按照“2.3.2”项方法制备绞股蓝(S1)提取液,按“2.1”项下色谱条件进样测定,分别于0、24、48 h 进样分析, 参照峰为绞股蓝皂苷XLIX 和皂苷A 峰, 分别计算2 个峰的相对保留时间RSD 和相对峰面积RSD。 结果显示,两个峰相对保留时间的RSD 为0.04%和0.05%, 相对峰面积的RSD 为0.53%和1.27%, 表明本次试验制备的供试品溶液在48 h 内较稳定。
2.6 重复性实验 采用加标回收实验的绞股蓝提取液6 份,按“2.1”项下色谱条件进样测定,参照峰为绞股蓝皂苷XLIX 和皂苷A 峰, 分别计算2 个共有峰的相对保留时间RSD 和相对峰面积RSD。 结果显示,两个峰相对保留时间的RSD 为0.07%和0.01%, 相对峰面积的RSD 为0.75%和0.60%,表明本次实验方法的重复性良好。
2.7 加标回收实验 精密称取绞股蓝供试品(S3),每份约0.2 g,分3 组,添加混合标准对照品的浓度水平为5、25 μg/mL 和200 μg/mL,每组6个平行,按“2.1”项下色谱条件进样测定,参照峰为绞股蓝皂苷XLIX 和皂苷A 峰,记录峰面积,计算两种皂苷3 个水平加标回收率为91.38% ~98.4%,RSD 小于2.48%,实验结果如表3 所示,表明本实验方法回收率较好。
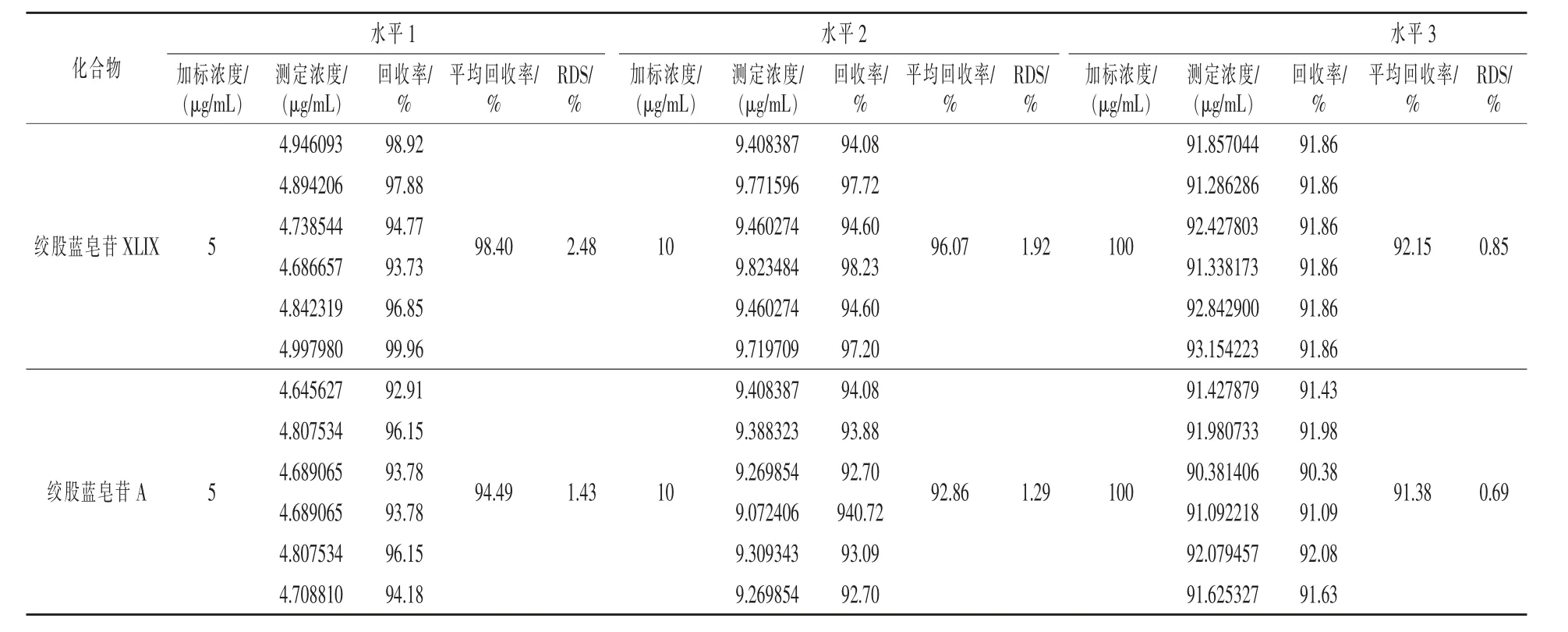
表3 加标回收率试验结果
2.8 样品含量测定 按“2.3.2”项分别对6 个样品进行制备,按“2.1”项下色谱条件进样测定,对从市面上购买的6 批样品进行测定, 结果表明样品S2、S3、S5 绞股蓝皂苷XLIX 和A 未检出,实验结果如表4 所示。
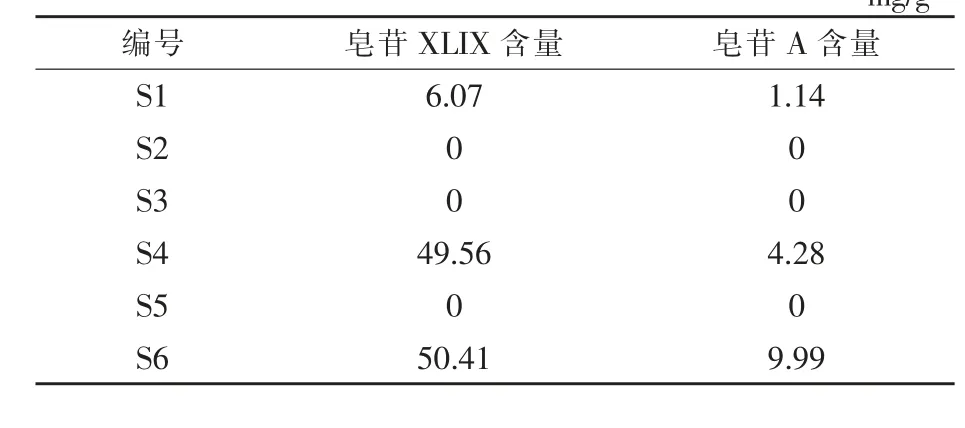
表4 含量测定结果 mg/g
实验测得的含量结果显示, 市场中购买的绞股蓝提取物中皂苷的含量差异较大, 质量参差不齐,这可能与绞股蓝不同的基原、品种和加工方式等有较大关系 (Zhang 等,2021;Zhang 等,2021;Dong 等,2018;Huang 和Yu,2000), 绞股蓝的不同部位和生长时期对皂苷种类和含量也有影响(乐圆,2010),有待进一步进行研究。
4 结论
本实验前处理利用甲醇直接提取法和SPE柱净化目标化合物, 并使用HPLC-UV 对绞股蓝皂苷XLIX 和皂苷A 进行准确的定量测定, 建立了SPE-HPLC-UV 检测方法。 本实验所建立的方法样品前处理简单、分析时间短、检测效率显著提高、其稳定性、精密度、重复性、线性关系良好,适用于绞股蓝提取物中对绞股蓝皂苷XLIX 和皂苷A 进行准确的定量。
猜你喜欢
中国粮油学报(2019年4期)2019-07-12
中成药(2018年9期)2018-10-09
中成药(2018年2期)2018-05-09
中成药(2018年1期)2018-02-02
中成药(2017年9期)2017-12-19
恋爱婚姻家庭·养生版(2017年12期)2017-12-07
恋爱婚姻家庭(2017年36期)2017-07-22
中成药(2017年6期)2017-06-13
中成药(2017年4期)2017-05-17
陕西画报(2016年1期)2016-12-01
