
转录起始位点预测方法的研究进展
2024-02-04 03:17郝瑞杰李星雨马子菡邵燕瑞晋朦朦杨稚萱杨柯华丁依菲李浩哲魏雪锋
信阳师范学院学报(自然科学版) 2024年1期
郝瑞杰,李星雨,马子菡,李 敏,邵燕瑞,晋朦朦,杨稚萱,杨柯华,丁依菲,李浩哲,魏雪锋
(信阳师范大学 生命科学学院, 河南 信阳 464000)
0 引言
mRNA转录起始位点(Transcription start sites,TSS)是基因组注释的关键内容。其上游为核心启动子,而TSS下游则是同样具有重要转录调控作用的5’非编码序列[1]。同时,紧靠TSS两侧的序列一般都是转录起始前复合物(Pre-initiation complex,PIC)的结合位点(图1)[2]。因此,TSS的确定是理解真核生物和原核生物基因表达和调控模式的基础[3]。但是,由于进化适应性等因素,导致不同品种/地域的动植物的TSS位点可能存在偏差。目前,可变启动子以及可变TSS位点已在多个研究中被证实[4-5]。基于实验方法的TSS鉴定存在着耗时、费力、昂贵、成功率低等诸多问题。如何克服这些问题,或者寻找一种折中的、可替代的TSS研究方法,是摆在研究人员面前的首要问题。在过去30年中,各种类型的预测程序相继被开发出来。它们可以高通量预测特定物种整个基因组的TSS信息,也可以为特定基因/实验提供候选TSS位点,以提高实验操作效率。基于此,本文在强调模型预测重要性的同时,重点分析了不同类型TSS预测软件的建模原理、特征采集、运行效果以及它们的各自优缺点,以方便后续研究人员的参考应用。

图1 真核生物转录起始位点与Ⅱ类启动子的结构关系图
1 TSS预测的必要性
1.1 TSS注释的意义
在真核生物中,核心启动子序列包括TATA盒、起始子 (Initiator element,Inr)、TFIIB识别元件(TFIIB recognition element, BRE)和下游启动子元件(Downstream promoter element, DPE)。调控转录起始频率的CAAT盒和GC盒属于上游调控元件(图1)[6]。目前研究表明这些常见启动子序列其实不足以启动基础转录。不同基因的核心启动子元件和近端顺式调控元件存在自身特异性,但都与TSS位点有着密切关系(图1)。因此,TSS的准确定位与注释是分析启动子元件位置与组成的前提[7]。
仅仅研究DNA一级结构的转录调节位点很难解释现有的表达调控现象,因为即使最常见的启动子基序也不是在所有基因的启动子序列中都存在。Inr是真核生物中启动子最常见的序列特征,但仅存在于53.3%的真核生物启动子中,而TATA box则仅在24.4%的真核生物启动子中被发现[6],同时,这些序列的组合在不同基序中也不尽相同[8]。缺乏一致的启动子基序导致人们猜测DNA二级结构可能是许多启动子转录的基础[9]。事实上,二级DNA结构,比如G-四联体[10]、富含胞嘧啶的嵌入基序结构(i-motif)[11]和十字形结构[12]等都可以影响核心启动子功能。 然而,DNA二级结构鉴定并不像基序或重复鉴定那么简单[13],准确的TSS鉴定及注释对于发现它们至关重要。
另外,可变TSS位点(或TSR)[5]以及最小启动子序列(核心元件的组合)[14]在国内外广受关注,该研究本身就是TSS注释的重要内容。
1.2 TSS实验鉴定法的不足
近年来,鉴定TSS和的实验方法取得了很大进展,也积累了大量与表达调控相关的序列数据[15-16],为进一步的功能研究和生物育种提供了广泛的资源[17]。
但实验成本很高、耗时、费力,且常常遭遇失败[18]。这主要是因为获得特定生物体全部基因的完整转录本的实验难度很大,这些序列的开头和结尾可能缺失或不完整[19-20]。而定位TSS需要完整的cDNA转录本,然后通过SIM-4[21]、BLAT[22]和GMAP[23]等比对工具将其定位到基因组中。一般来说,序列组装,TSS定位的准确度与软件的匹配算法、序列的长度和非翻译区域的覆盖范围有关(特别是在5’UTR区域)[19-20]。
因此,鉴于实验鉴定的诸多缺点,对单个基因[3]和整个基因组[24]的TSS位点进行精确计算定位,正在成为基因组学和后基因组学研究的重要内容之一。
2 TSS位点预测工具的发展及分类介绍
现有预测工具的分类方法有很多,根据预测目标可分为2类:1)预测已知基因TSS上游启动子区域;2)专注于寻找TSS位点[25]。根据算法可分为3类:1)基于得分函数的方法;2)传统的机器学习算法;3)基于深度学习的方法[26]。表1列举了部分主流的TSS预测软件,并分组说明了不同软件的优缺点以及应用物种。表1中对软件的分组主要是依据其所采集的序列特征信息,同时结合了算法,相较之前的分类,更为细致和清晰。文中在详细介绍不同类型软件的运行原理、性能及适用范围的同时,还重点分析了序列特征信息及算法革新对软件运行效果的影响。
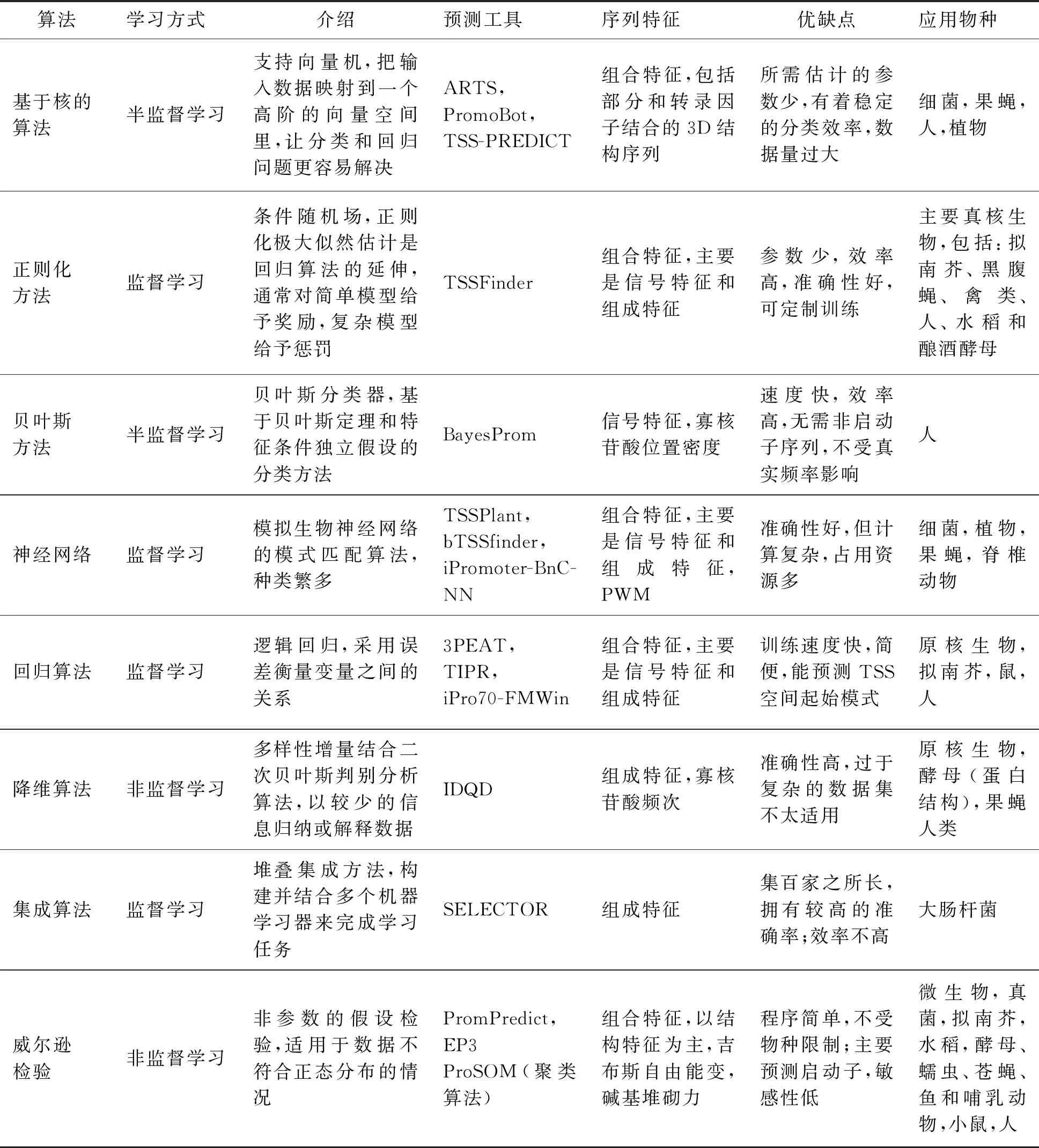
表1 TSS预测的主要工具/算法
2.1 早期依赖于启动子识别的预测方法
鉴于TSS位点紧邻启动子的核心区域下游,早期的TSS 定位基本上依赖于对应启动子识别的结果。识别方法可大致分成两类:基于核苷酸组成(六聚体频率)的方法[27-29]和基于信号(已知基序的富集度)的方法[30-32]。前者主要利用了启动子序列与背景序列在全局碱基组成上的差异,因此仅限于判断待定序列是否属于启动子区域,无法给出精确位置信息,也无法对TSS 进行定位。后者则通过发现启动子区域内特异的局部保守模式进行识别,可以对启动子的位置进行预测,并将识别结果特定距离的DNA位点近似作为TSS[33]。这些方法的主要优点是分析的性状少、模型简单。近年来,在lncRNA的研究中,仍在被相关人员使用。张芳芳等[34]利用对TATA-box 和CAAT-box定位,确定了玉米胚萌发阶段杂种优势相关miRNA 的编码MIR 基因的转录起始位点。
需要指出的是,由于基因调节元件的或然性、可变性和基序组合的不确定性以及调节网络的复杂性,早期方法的识别特异性与计算精度普遍较低[30,35]。BAJIC等[36]于2004年测试了8个主要的启动子预测模型,发现没有一个程序同时使得敏感性和阳性预测值高于65%。
2.2 基于组成合并信号特征的预测方法
预测算法的准确性取决于以下3个因素:1)训练和测试数据集的质量;2)用于区分启动子和非启动子序列的特征;3)数据模型(分析算法)是否科学、合适[25,37-38]。为了提高预测的精准度,研究人员对识别算法进行了大量的优化和改进[25,39],并直接用于TSS鉴定分析。依据辨识度的强弱,对启动子的组成特征、信号特征和二维结构特征等进行加权、筛选,然后构建预测模型。比较典型的有逻辑回归[40-41]、支持向量机(Support vector machines,SVMs)[42]、线性链条件随机场模型(Linear chain conditional random fields, LCCRFs)[38]、贝叶斯分类[40]、多样性增量结合二次判别分析的IDQD算法(Increment of diversity with quadratic discriminant analysis, IDQD)、降维算法与贝叶斯网络的结合[43-44]和神经网络[37,45]等。
逻辑回归属于分类模型,算法简单,但预测效果并不差。其本质是假设数据服从这个分布,然后使用极大似然估计做参数的估计。比较知名的两个预测软件是3PEAT[40]和TIPR[41],它们不能直接输出TSS位点,通常都输出,给定大小为8000~10 000核苷酸的序列以及一系列与TSS信号强度相关的信号峰值。这两个软件的区别在于:1)目标生物不同,3PEAT用于拟南芥模型,TIPR用于小鼠模型;2)TSS位点预测的峰形分类不同,TSS-3PEAT为窄峰、宽峰和弱峰,而TIPR则为单峰和宽峰。
SVMs是在位点权重矩阵的基础上,通过设计函数集的某种结构,使每个子集中都能够取得最小的经验风险(如使训练误差为0),然后选择适当的子集使置信范围最小,则这个子集使经验最小的函数便是最优函数,从而实现结构风险最小化[46]。ARTS是通过使用复杂的字符串核所开发的一种更为高效的SVM训练和评估算法, 与当时最先进的TSS预测方法相比,该方法实现的高达两倍的真阳性率。同样地,应用SVM技术,AZAD等[47]开发了一种新的PromoBot工具,其灵敏度达到了89%,特异性为86%。
条件随机场(Conditional random fields, CRFs)已经被成功地用于基因结构的表征[48-50],但基于CRF的模型并没有直接用于寻找TSS信号。LCCRFs是在一般条件随机场(CRFs) 基础上增加了限制,其在推理算法的速度上有明显的优势[48]。TSSFinder是一种利用LCCRFs预测注释基因TSS的新方法[38]。该方法直接从蛋白编码基因中寻找集中的TSS信号,并可从离散TSS信号基因中,确定离起始密码子最近的TSS信号。
贝叶斯分类[40]主要是利用核心启动子区六聚体(如TATA-box、Inr等基序)的位置密度建模,来检测人类启动子序列的TSS。该计算方法的优势在于运行速度快,效率高,但是六聚体没有固定组合及位置,该生物信号的不确定性会干扰该模型标记DNA序列的效能。IDQD是在贝叶斯分类的基础上,结合了降维算法(即ID算法结合QD算法),其理论基础是普遍的信息最大化原理,即每一类特征用ID算法将特征信息向高维空间投影,以期获得该类特征更为细致的信息,不同类特征则分别构建不同的ID,用这些ID构成一个判别向量,采用非线性判别函数QD来进行分类判别。多个研究表明,IDQD算法在应用到TSS位点鉴定时,其效果与当前性能最好的SVM算法相当[51]。
神经网络是多层的非线性模型,首先用Boltzman Machine(非监督学习)学习网络的结构,然后再通过Back Propagation(监督学习)学习网络的权值。TSSPlant[25]和TransPrise[37]都采用了神经网络。尤其是TransPrise,使用卷积神经网络来提高神经网络的模型的预测性能。除了以上6种常用算法之外,还有图像处理技术(IBBP)[52]和堆叠集成方法(SELECTOR)[53]等。这些预测软件在被提出的原始文献里,都声称该算法达到了很高的精确度与特异性,也确实与当时的知名算法进行了对比分析。但在引证文献的研究中,其计算性能可能会有差别。事实上,这些算法各有优缺点,其在不同研究中使用效果的差异,可能和数据样本、物种特征、地域环境以及开发时间有关系。
2.3 包含5’UTR特征信息的预测方法
具有重大贡献度的TSS特征的筛选、加权是影响各种模型预测准确度的因素之一。TSS-TLS 距离作为转录起始的一个重要特征,一直以来被人们所忽视。事实上,TSS-TLS 距离大小(本质上包括5’UTR区域和对应的内含子)具有一定分布规律。植物单子叶和双子叶的5’UTR区长度一般在650~950个核苷酸之间[54],人类和小鼠的5’UTR区长度一般在500~1000个核苷酸之间[55],因此起始密码子与TSS之间的假定距离通常在500~1500之间。在原核生物中,TSS信号位于距离基因起始处100 nt处,但在某些情况下,这一距离可能接近400 nt[42]。研究人员认为可以根据TSS-TLS 距离的经验概率分布,建立离散经验分布函数,对预测得分进行修正。杜耀华等[33]利用该方法对大肠杆菌真实序列数据进行了测试,结果表明该定位算法可实现对真实转录起始位点位置的有效预测,与已有算法相比,当敏感性指标同为0.85左右时,特异性指标可从0.20提高至0.65,提高了定位准确率。黎志凤等[56]在该预测方法指导下,成功鉴定了黏球菌Myxococcus DK1622 来源的双拷贝GroELs基因的TSS,但同时指出预测结果需实验修正。EUGENIYA等[57]采用基于期望最大化和神经网络分类方法的TSSPlant算法,结合UTR长度的概率分布对4种基因组组装和注释并不完整的针叶树TSS位点进行了预测,其预测结果的准确性得到了DNA双链的标准自由能谱、CG偏态分布及TFBS位置分布的进一步证实。TSSFinder更是将TSS-TLS 距离这一特征变量直接构建到了LCCRF模型中,虽然该预测模型仅包含了4种类型的特征信息(假定的TATA-box位置(如果有的话)、TSS到TATA-box的距离、TSS到起始密码子的距离以及TSS区域的组成),却比以前发布软件具有更高的准确性[38]。
2.4 基于空间结构特征的预测方法
一般来说,信号特征(PWMs、启动子元件基序及其相距离、TFBS密度和NFR区域等)和组成特征(寡核苷酸频次、CG偏态、AC偏态、GC3富集、遗传变异差异、CpG岛和表观遗传标记等)常常被用来构建预测模型。然而,启动子区域与非启动子区域在DNA稳定性[58-59]、可弯曲性、曲率等[60-61]空间结构特征方面同样存在明显差异。DNA稳定性与转录起始前双链分子的熔解有关,通常用DNA双链的标准自由能来计算。该方法已成功地应用于多种真核生物的启动子鉴定[62]。另外,多个文献报道在TSS上游存在弯曲DNA区域,而与DNA结合蛋白相互作用的序列则呈现了更高的弯曲性[60,63]。PromPredict是一款典型的基于TSS相邻区域之间的稳定性差异而开发的软件[39,62],它成功预测了913个微生物基因组序列的启动子区域,并且发现启动子区域与其下游区域的平均自由能值不同,这取决于它们的GC含量。事实上,DNA序列的空间结构特征与其二维的核苷酸组成,相互联系,密不可分。BONDAR等[57]建议在对非模式植物数据进行分析的时候,可以将基于组成特征与信号特征的TSSPlant与基于空间结构特征的PromPredict算法一起使用,以提高TSS的预测精度。
2.5 预测法-实验法联合分析
虽然基于生物信息学分析的预测法,在算法上以及特征信息的提取上都得到了极大程度的发展,但预测误差仍时常出现。同时,预测的假阳性结果中有些可能是潜在的备选TSS 位点,这些都需要实验方法来分析鉴定。预测结果作为参考和指导,实验结果作为验证和支撑,预测法与实验法相辅相成,被认为是更为理想的TSS鉴定模式。张芳芳等[34]在预测了玉米miRNA 编码基因MIR的TSS之后,又用5’RACE 技术进行了分析验证,在证明预测方法可行性的同时,也进一步提高了所鉴定TSS位点的可信度。黎志凤等[56]应用遗传片段分析技术内标法结合正态分布理论,对预测结果进行了修正,展现了预测联合实验TSS鉴定技术方案的巨大性能。
3 展望
预测的TSS及其推测的启动子区域可为后续的实验研究提供参考与指导,并为更好地理解基因调控和物种之间的进化关系提供了宝贵的资源。TSS鉴定可用于分子遗传辅助育种和基因组编辑,为更精确地定位与生长、抗逆以及抗病等适应性性状相关的功能基因组区域(QTL)和QTN提供了机会。
由于基因调控的自然多样性以及启动子序列信息的复杂性,预测启动子及TSS其实是一项非常困难的任务。随着分子生物学以及高通量测序技术的不断发展,人们对转录组及其调控的理解将更加深入,当前算法中尚未考虑的相关信息在新的计算方法中也会被不断识别。未来新模型的预测性能将会得到进一步提升。其发展方向有可能具备以下3点:1)新模型所覆盖的启动子鉴别特征信息精简,但辨识度高,贡献度大;2)新算法简便、高效、精准;3)区别种属特异性和/或种内组织特异性特征(尽管有些研究认为混合不同物种序列似乎不会降低预测的准确性)。现阶段,具体到某一模型的构建或基因组序列的TSS鉴定,模型预测结合实验验证仍是首选的方法。
猜你喜欢
新医学(2023年10期)2023-12-09
黄河之声(2022年10期)2022-09-27
中学生数理化(高中版.高二数学)(2022年4期)2022-05-25
中学生数理化(高中版.高二数学)(2022年4期)2022-05-25
南方医科大学学报(2022年3期)2022-04-13
上海金属(2021年6期)2021-12-02
昆明医科大学学报(2021年3期)2021-07-22
浙江大学学报(农业与生命科学版)(2021年3期)2021-07-10
生物学通报(2019年3期)2019-02-17
中学生数理化·八年级物理人教版(2017年11期)2017-04-18
