
昂拉地韦潜在基因毒性杂质的合成、表征和检测
2024-01-23 06:05刘呈武刘卓伟黎志豪
合成化学 2024年1期
李 波, 刘呈武, 刘卓伟, 黎志豪.
(广东众生睿创生物科技有限公司 工艺开发部, 广东 广州 510700)
流行性感冒是由流感病毒引起的一种急性呼吸道传染病,其中甲型流感病毒的传染性最强且变异率较高[1]。流感病毒RNA聚合酶中的PB2蛋白可以识别并结合宿主mRNA的帽状结构,产生病毒mRNA,进而合成病毒蛋白[2]。昂拉地韦的化学名称为(2S, 3S)-3-[[6-环丙基-5-氟-2-(5-氟-1H-吡唑并[3,4-b]吡啶-3-基)嘧啶-4-基]氨基]二环[2.2.2]辛烷-2-羧酸,是由广东众生睿创生物科技有限公司研发的全新结构的RNA聚合酶PB2蛋白抑制剂[3-7]。昂拉地韦是具有明确作用机制和全球自主知识产权的一类创新药物,临床上拟用于成人单纯性甲型流感的治疗,是国内首个获批临床试验的治疗甲型流感的小分子RNA聚合酶PB2蛋白抑制剂,可以抑制病毒生命周期基因组的转录和复制等多种功能,达到抗甲型流感病毒的作用。目前,昂拉地韦已完成III期临床研究,2023年7月,众生睿创公布III期临床试验顶线数据,昂拉地韦较安慰剂可显著缩短中位7项流感症状缓解时间和中位发热缓解时间,较安慰剂或对照药可更好更快地降低甲型流感病毒载量,显著缩短病毒转阴时间,现推进NDA上市申请中。昂拉地韦的结构式如图1所示。

图1 昂拉地韦的结构
昂拉地韦原料合成工艺中用到物料亚硝酸叔丁酯,存在产生工艺杂质SMI-H[8]的风险,该杂质中含有基因毒性警示结构叠氮基,为潜在的基因毒性杂质,能够直接或间接作用于人体DNA,导致基因突变或诱发疾病[9-16]。本文参照人用药品技术要求国际协调理事会(ICH)M7遗传毒性杂质指导原则,采用基于专家知识规则(Derek Nexus 6.0.1)和统计学模型(Sarah Nexus 3.0.0)的2个软件对SMI-H进行预测,结果显示其为风险较高的潜在基因毒性杂质(Derek预测结果为Plausible, Sarah预测结果为Positive)。基于基因毒性杂质对人体有较大的安全隐患,为保证昂拉地韦药物的安全性,需要对该杂质进行充分的研究。由于SMI-H是未经报道的全新结构分子化合物,因此通过检索相关文献[17-19],自行设计路线来定向合成潜在基因毒性杂质SMI-H(图2)。
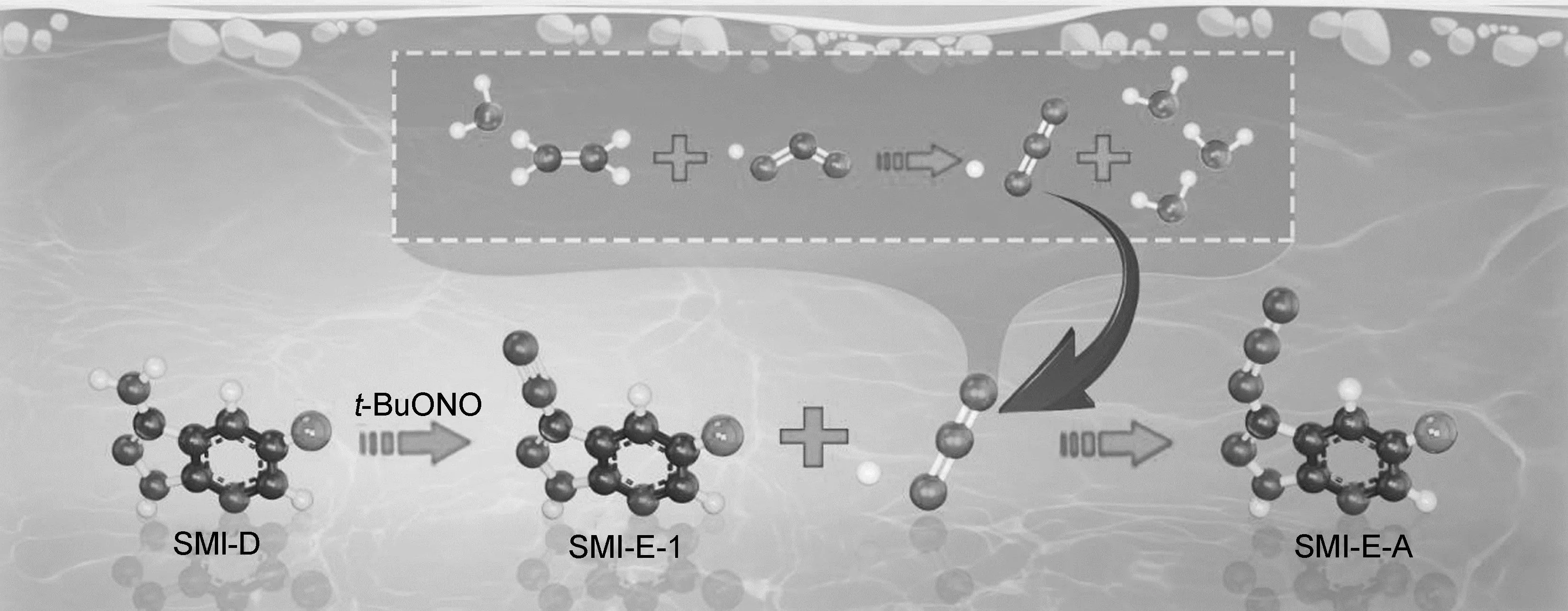
图2 化合物SMI-E-A的合成机理
1 实验部分
1.1 仪器与试剂
AVANCE Ⅲ 400 MHz型核磁共振波谱仪(DMSO-d6为溶剂,TMS为内标,德国Bruker有限公司); AB Sciex API 150型质谱仪(美国ABI公司); 安捷伦1260 Infinity II型高效液相色谱仪; AB SCIEX Triple Quad 4500型液质联用仪。
所用试剂均为市售分析纯。
1.2 合成
(1) 化合物2的合成
取2-氯-5-氟烟腈(1)(12.54 g, 80 mmol)置于500 mL三口烧瓶中,加入无水正丁醇(50 mL)搅拌。用恒压滴液漏斗缓慢加入质量分数为50%的水合肼(32.09 g, 320 mmol),反应体系升温至100 ℃搅拌反应6 h。反应完毕,将反应体系降温至30 ℃,控温搅拌2 h。过滤,滤饼用水(100 mL)和正庚烷(50 mL)分别淋洗1次,控制内温在60 ℃以下,减压浓缩至无馏分。用真空干燥箱减压干燥至恒重,得到浅黄色固体210.15 g,收率83.29%,纯度99%;1H NMR(600 MHz, DMSO-d6)δ: 12.08(s, 1H, NH), 8.38(dd,J=2.7 Hz, 1.8 Hz, 1H, ArH), 7.98(dd,J=8.8 Hz, 2.8 Hz, 1H, ArH), 5.56(s, 2H, NH2);13C NMR(150 MHz, DMSO-d6)δ: 154.43, 152.84, 150.11, 148.73, 148.70, 138.29, 138.10, 115.01, 114.87, 105.79, 105.74; DEPT135(150 MHz, DMSO-d6)δ: 138.29, 138.10, 115.01, 114.87; MS(ESI)m/z: Calcd for C6H5FN4{[M+H]+}153.13, found 153.0。
(2) 3-叠氮基-5-氟-1H-吡唑[3,4-b]吡啶的合成
取3-氨基-5-氟-1H-吡唑[3,4-b]吡啶(2)(10.0 g, 65.7 mmol)置于500 mL三口烧瓶中,加入无水乙腈(100 mL)搅拌。用恒压滴液漏斗缓慢加入亚硝酸叔丁酯(10.2 g, 98.6 mmol)和50%的水合肼(9.9 g, 98.6 mmol),反应体系升温至60 ℃搅拌反应4 h。反应完毕,将反应体系降温至30 ℃,减压浓缩至无馏分,柱层析(洗脱剂:石油醚 ∶乙酸乙酯=10 ∶1,V∶V;二氯甲烷 ∶甲醇=10 ∶1,V∶V)纯化,得到白色固体(3)10.2 g,收率87.18%,纯度97%;1H NMR(600 MHz, DMSO-d6)δ: 13.69(s, 1H, NH), 8.62(dd,J=2.8 Hz, 1.8 Hz, 1H, ArH), 8.01(ddd,J=8.3 Hz, 2.8 Hz, 0.7 Hz, 1H, ArH);13C NMR(150 MHz, DMSO-d6)δ: 155.67, 154.05, 149.82, 140.84, 140.64, 139.51, 139.48, 113.83, 113.69, 106.17, 106.12; MS(ESI)m/z: Calcd for C6H3FN6{[M-H]+}177.13, found 177.0。
(3) 化合物SMI-H的合成
向500 mL反应瓶中加入无水二氯甲烷(200 mL, 20 v/w), 化合物3(10.0 g, 56.2 mmol),开启搅拌,加入3,4-二氢吡喃(4.96 g, 59.0 mmol)和对甲基苯磺酸(1.93 g, 11.2 mmol), 25 ℃搅拌反应24 h。向反应液中加入饱和碳酸氢钠溶液(200 mL, 20 v/w),在25 ℃下继续搅拌10 min后静置,分液,有机相用100 mL饱和氯化钠水溶液洗涤,无水硫酸钠干燥,过滤,减压浓缩,干法上柱。流动相:正庚烷 ∶乙酸乙酯=1 ∶0~10 ∶1,V∶V。浓缩后,30~40 ℃真空烘箱干燥,得到白色固体SMI-H 11.2 g,收率76%,纯度99.1%[HPLC面积归一化法:色谱柱ACE Excel 3 C18-PFP(4.6 mm×150 mm, 3 μm),流动相A为纯化水,流动相B为乙腈,梯度洗脱,检测波长为210 nm,柱温35 ℃,流速1.0 mL·min-1,tR=14.603 min];1H NMR(600 MHz, DMSO-d6)δ: 8.70(s, 1H, ArH), 8.11(dd,J=10.0 Hz, 2.5 Hz, 1H, ArH), 5.97(dd,J=10.0 Hz, 5.0 Hz, 1H, ArH), 3.94(d,J=10.0 Hz, 1H, CH), 3.68(dt,J=20.0 Hz, 5.0 Hz, 1H), 2.40(tdd,J=12.9 Hz, 10.6 Hz, 4.2 Hz, 1H), 2.04~2.00(m, 1H), 1.92(dd,J=13.0 Hz, 2.9 Hz, 1H), 1.76(tdd,J=10.5 Hz, 7.4 Hz, 3.4 Hz, 1H), 1.60~1.54(m, 2H);13C NMR(150 MHz, DMSO-d6)δ: 156.28, 154.65, 148.13, 140.98, 140.78, 139.68, 139.64, 114.64, 114.50, 107.61, 107.56, 82.34, 67.62, 29.19, 25.06, 22.80; DEPT135(150 MHz, DMSO-d6) CHδ: 140.98, 140.78, 114.65, 114.50, 82.34; CH2δ: 67.62, 29.19, 25.06, 22.80; DEPT90(150 MHz, DMSO-d6) CHδ: 140.98, 140.78, 114.65, 114.50; MS(ESI)m/z: Calcd for C11H11FN6O{[M+H]+}263.25, found 263.10。
1.3 液质联用法
液相色谱:检测器-AB SCIEX Triple Quad 4500;色谱柱:飞诺美Kinetex C18柱;流动相A:甲酸 ∶水(1 ∶1000);流动相B:甲醇;柱温:40 ℃;进样体积:5 μL;采集时间:10 min;洗针液:体积分数为50%的甲醇-水;洗脱程序:梯度洗脱;质谱条件:扫描类型-MRM;离子化形式:ESI;极性:positive; CAD(碰撞气压力):8 psi; GS1(雾化气压力):50 psi;GS2(雾化气压力):50 psi; CUR GAS(气帘气压力):40 psi; IS(离子化气压):5500 V; Temp(干燥气温度):500 ℃;阀切换程序:0~4 min, To Waste; 4~8 min, To MS; 8~10 min, To Waste。
2 结果与讨论
2.1 化合物2的工艺条件优化
以 2-氯-5-氟烟腈5 g的反应规模,分别考察了溶剂种类、水合肼的用量和反应温度对化合物2收率和选择性的影响。选择性为反应结束时体系中化合物2的含量,采用面积归一化法HPLC进行分析。
(1) 溶剂的种类
表1研究了不同溶剂体系(4 v/w)对反应选择性的影响。由表1可见,醇类溶剂体积的反应选择性明显优于二氯甲烷体系,乙醇和正丁醇的反应选择性优于甲醇体系,其中以正丁醇的选择性最佳,因此,选择正丁醇作为反应溶剂。

表1 溶剂种类对化合物2反应选择性的影响
(2) 水合肼的用量
表2研究了不同水合肼用量对反应选择性的影响。由表2可见,4.0 eq和5.0 eq的水合肼体系选择性最高,由于水合肼具有强碱性和吸湿性,本着提质增效的原则,选择4.0 eq的水合肼。

表2 水合肼用量对化合物2反应选择性的影响
(3)反应温度
表3研究了反应温度对收率的影响。由表3可见,温度为95 ℃和105 ℃时收率最高,温度降低时,收率亦会降低。
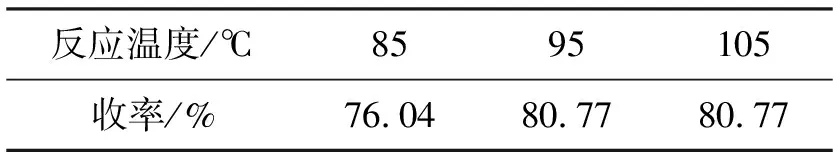
表3 反应温度对化合物2收率的影响
2.2 化合物3(SMI-E-A)的潜在反应机理解析
经文献检索,未见有SMI-H的制备方法,分析SMI-E-A的叠氮化反应机理(图2),水合肼和亚硝酸类似物生成叠氮酸,SMI-D在亚硝酸叔丁酯的作用下生成重氮盐中间态,叠氮酸进攻重氮盐中间态生成SMI-E-A。
2.3 SMI-H在起始物料和昂拉地韦原料药中的方法开发和检测
根据ICH M7指导原则,基因毒性杂质的可接受风险摄入量毒理学阈值(TTC)为每天不超过10 μg。昂拉地韦每日服用的最大剂量为800 mg,按照TTC公式(限度=10 μg/日最大剂量)计算,SMI-H在原料药和片剂中的可接受限度定为12.5×10-6。
采用自行开发的高效液相色谱法对昂拉地韦关键起始物料SMI进行检测。经验证,该方法灵敏度高、专属性好、耐用性强且专属度高。具体色谱条件为:用十八烷基硅烷键合硅胶为填充剂(ACE Excel 3 C18-PFP, 150 mm×4.6 mm或效能相当的色谱柱);以水为流动相A,乙腈为流动相B,进行线性梯度洗脱;流速为1.0 mL/min;检测波长为210 nm;柱温为35 ℃;进样体积10 μL。
检测结果显示,SMI-H在多批次关键起始物料SMI检测中均有检出,检出数据范围为0.1%~0.2%,高于基于摄入量毒理学阈值(TTC)计算得到的限度12.5×10-6,低于SMI质量标准中的限度0.3%,典型谱图见图3。
图3 SMI-H液相检测数据
采用自行开发的液质联用法(LC-MS)对昂拉地韦原料药进行检测,该方法从系统适用性、专属性、线性及范围、检测限、定量限、准确度、精密度、稳定性和耐用性等方面进行验证,结果均符合《中国药典》2020年版四部“9101药品质量标准分析方法验证指导原则”的规定要求,因此可以运用此方法准确检测昂拉地韦原料药中SMI-H的含量。
检测结果如表4所示,SMI-H在多批次昂拉地韦原料药检测中均未检出,低于基于摄入量毒理学阈值(TTC)计算得到的限度12.5×10-6,III期临床批次原料药典型检测数据如表4所示,均未检出。
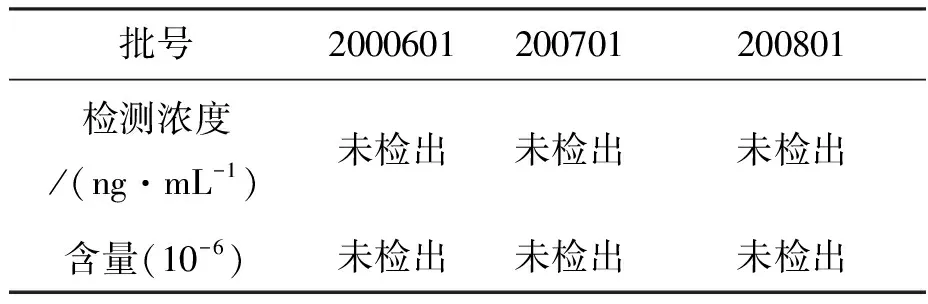
表4 昂拉地韦中SMI-H的多批次检测数据
2.4 SMI-H晶型稳定性
根据杂质对照品储存和多批次原料药检测的需要,本文同步考察SMI-H的晶型稳定性。0个月、 3个月、 6个月和12个月的XRD叠加谱图如图4所示,可以看出,SMI-H的晶型较稳定,角度峰一致,室温即可长期保存,为后续起始物料和原料药的批次检测提供有益的数据支持。
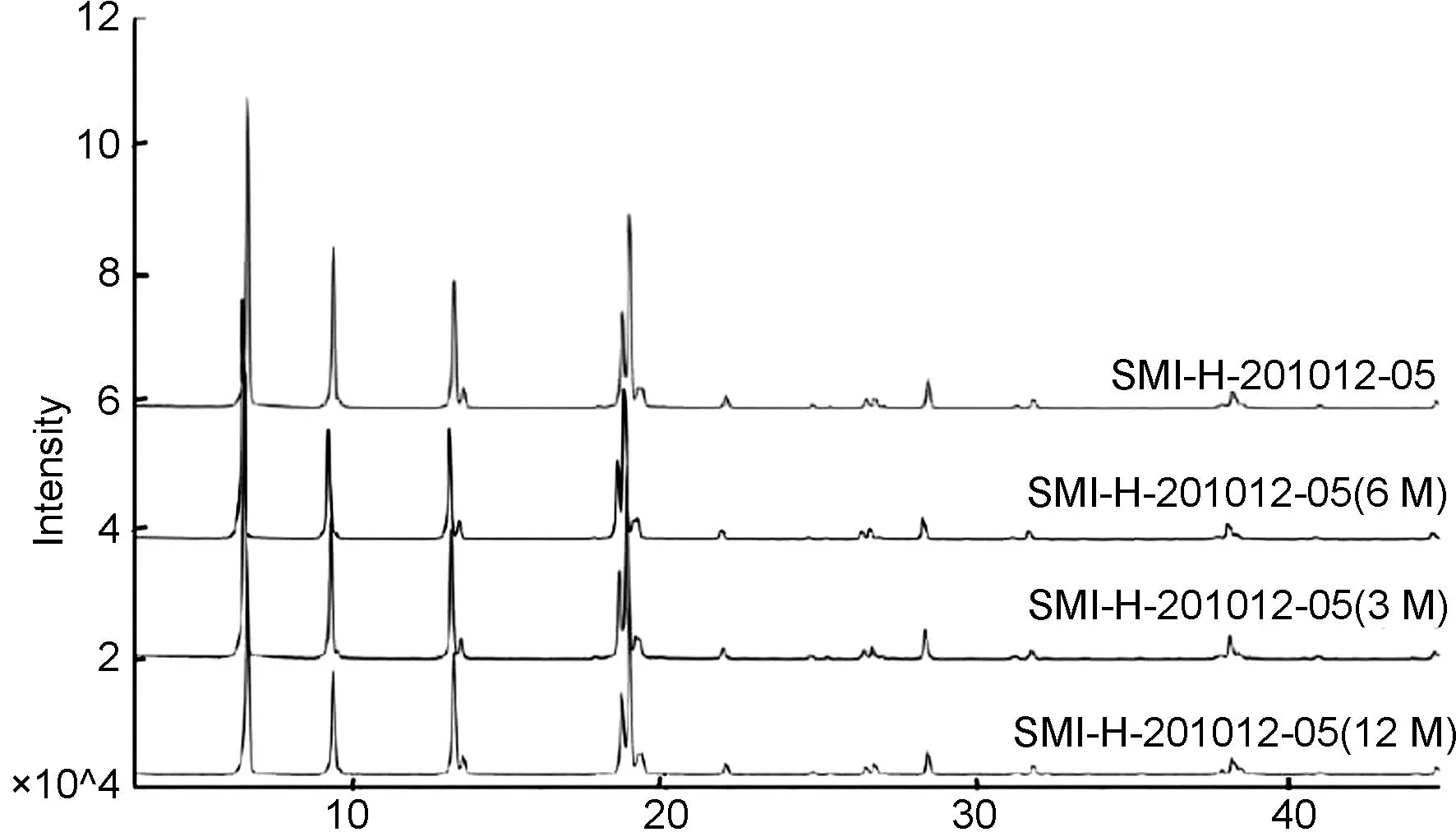
2θ/(°)
本文对昂拉地韦关键起始物料和原料药中潜在的基因毒性杂质SMI-H进行合成和表征,主要创新之处在于避免使用剧毒和易爆的叠氮化钠,合成工艺条件温和、原料易得并且操作简便。基于保证昂拉地韦商业化生产批次工艺性能和产品质量的考量,采用自行建立并验证的高效液相色谱和液质联用方法,对昂拉地韦关键起始物料和原料药中的SMI-H进行检测,数据显示在昂拉地韦关键起始物料有检出但低于SMI现行质量标准限度范围(≤0.3%),在昂拉地韦原料药中未检出,安全风险可控。本研究为昂拉地韦药品的质量控制、安全性评价及NDA申报上市提供数据支撑和有益参考,具有较高的实际应用价值。
猜你喜欢
氯碱工业(2022年6期)2022-11-21
云南化工(2021年7期)2021-12-21
化工环保(2021年2期)2021-04-25
艺术品鉴(2020年6期)2020-12-06
云南化工(2020年5期)2020-06-12
盐科学与化工(2019年11期)2019-12-04
中国盐业(2018年20期)2019-01-14
领导文萃(2017年6期)2017-03-24
中学生数理化·高一版(2016年7期)2016-12-07
中学生数理化·中考版(2015年12期)2015-09-10
