
新型抗菌药G0775的合成新方法
2024-01-23 06:04赵文豪杨玉社张银勇
合成化学 2024年1期
赵文豪, 杨玉社, 孟 昕, 张银勇*.
(1.南京中医药大学 新中药学院,江苏 南京 210023; 2.中国科学院 上海药物研究所,上海 201203)
抗生素的过度使用和滥用导致细菌耐药性迅速发展,这严重威胁了人类的健康,尤其是多重耐药(Multi-drug resistance, MDR)和广泛耐药(Extensively drug resistant, XDR)革兰氏阴性菌引起的感染,已成为全球卫生机构面临的主要挑战[1-4]。然而,具有全新结构和全新作用机制的抗生素研发工作却相对滞后,这进一步助长了细菌耐药性的出现和蔓延[5-7]。G0775(图1),化学名为(4S,7S,10S)-10-(S)-4-氨基-(2-(2-(4-((叔丁基)苯基)-4-甲基嘧啶)-5-甲酰胺)-N-甲基丁酰胺)-16,26-双(2-氨基乙氧基)-N-(氰甲基)-7-甲基-6,9-二氧代-5,8-二氮杂-1,2(1,3)-二苯环癸烷-4-甲酰胺[2],是由美国基因泰克PETER等研究人员通过对阿龙霉素结构改造发现的新型细菌信号肽酶I(Signal peptidaseI)抑制剂,结构新颖,作用机制独特,对典型的临床分离革兰氏阴性菌(鲍曼不动杆菌、大肠杆菌、肺炎克雷伯菌和铜绿假单胞菌)具有强大的体内外抗菌活性,且对MDR和XDR病原体敏感[8-10],证明了其在应对多药耐药革兰氏阴性菌日益增长的临床威胁方面有着巨大的潜力。

图1 G0775 的结构
G0775的文献合成方法是以(S)-2-氨基-2-(4-羟基苯基)乙酸(I-7)为起始原料,经碘代、亲核取代、Suzuki-Miyaura偶联、缩合、水解和脱保护等[11-14]共22步反应制得目标产物G0775(图2~3),总产率1.40%,中间体I-6是以(叔丁氧基羰基)-L-酪氨酸甲酯(I-1)为起始原料,经甲基化、碘代和亲核取代等共5步反应得到,产率为63.61%。该方法最大缺点为线性路线步骤太长,操作繁琐,总产率很低。
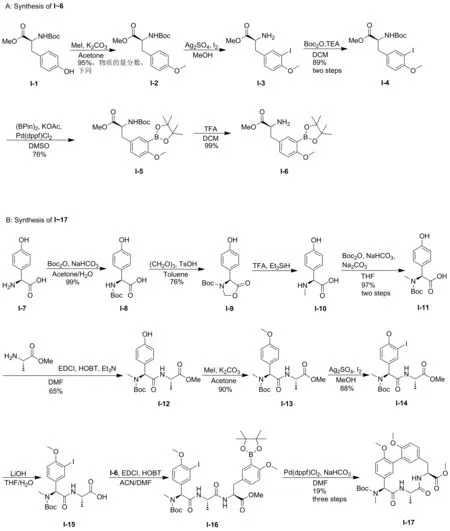
图2 G0775的原合成路线(A和B)
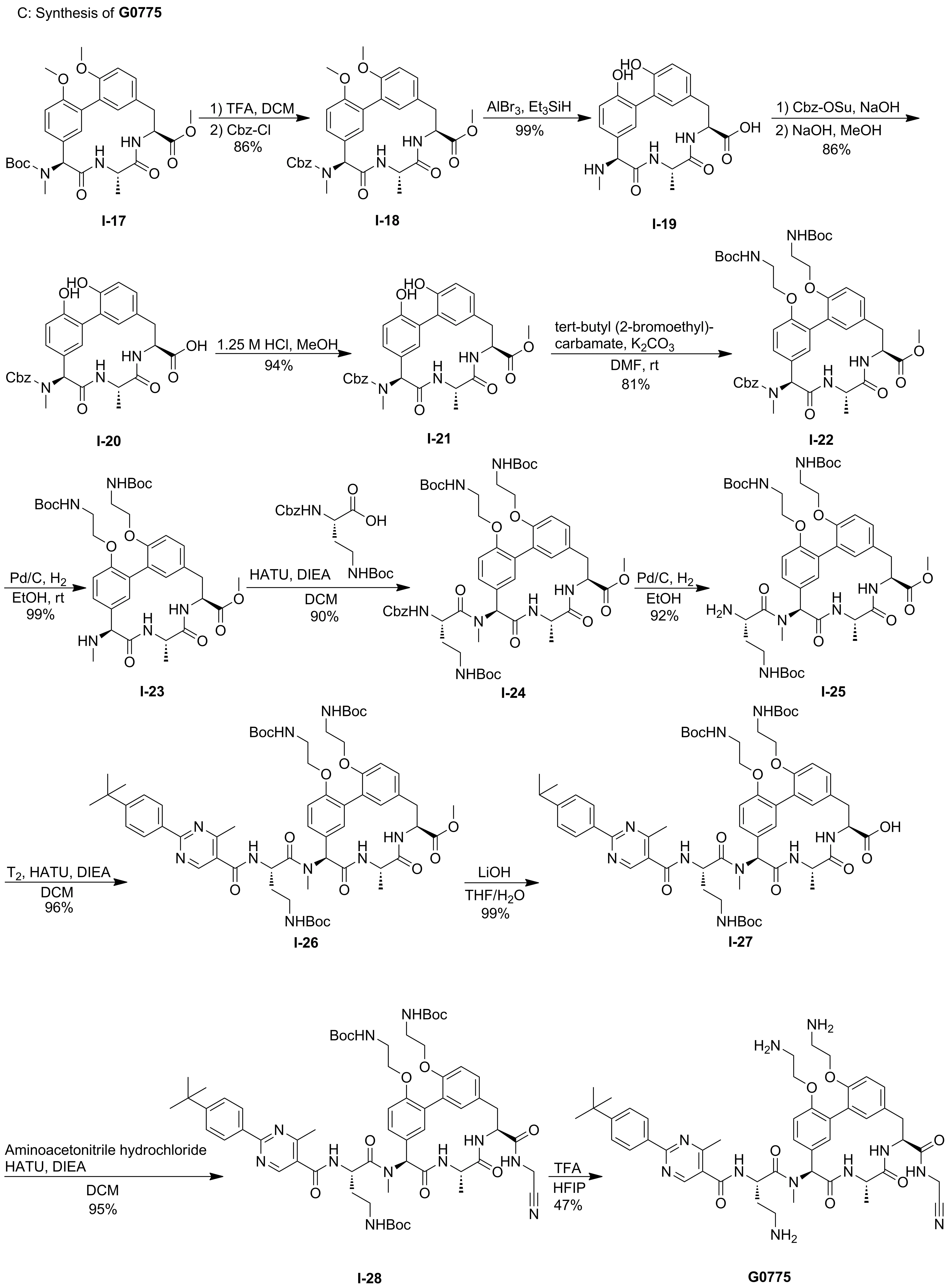
图3 G0775的原合成路线(C)
为解决上述路线存在的问题,本文重新设计了汇聚式的G0775的合成路线(图4~5:如图4所示,按文献方法[15-17],(S)-2-氨基-2-(4-羟基苯基)乙酸(I-7)经过碘代、亲核取代、Suzuki-Miyaura偶联和缩合等共7步反应得到中间体甲基(4S,7S,10S)-26-苄氧基-10-((((苄氧基)羰基)甲基)氨基)-16-羟基-7-甲基-6,9-二氧代-5,8-二氮杂-1,2(1,3)-二苯环癸烷-4-羧酸甲酯(I-40),中间体I-33是以L-酪氨酸(I-29)为起始原料,经碘代,亲核取代等共4步反应得到。本文设计以I-40为起始原料,经催化氢化、两步亲核取代反应得到中间体I-22,然后经脱保护,与(S)-4-((叔丁氧基羰基)氨基)-2-(2-(4-(叔丁基)苯基)-4-甲基嘧啶-5-甲酰氨基)丁酸(T4)(图6)缩合,再经水解和缩合反应,最后氯化氢脱除Boc保护,共8步得到目标产物G0775(图5)。本研究设计的新路线明显更具收敛性,比文献方法缩短7步反应,且操作更简便,总产率大幅度提高,为该类分子的合成研究提供有益参考。
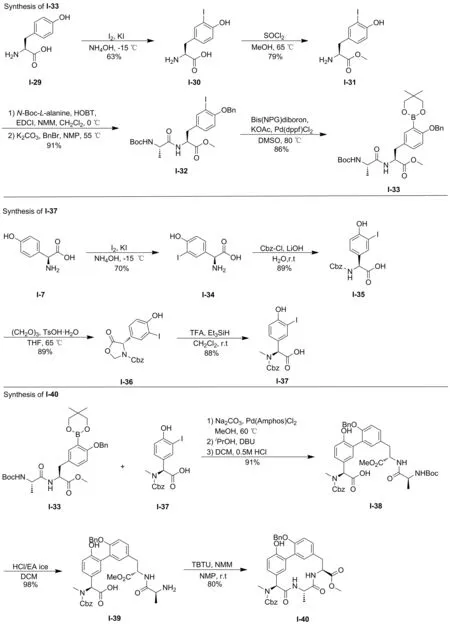
图4 大环中间体I-40的合成路线
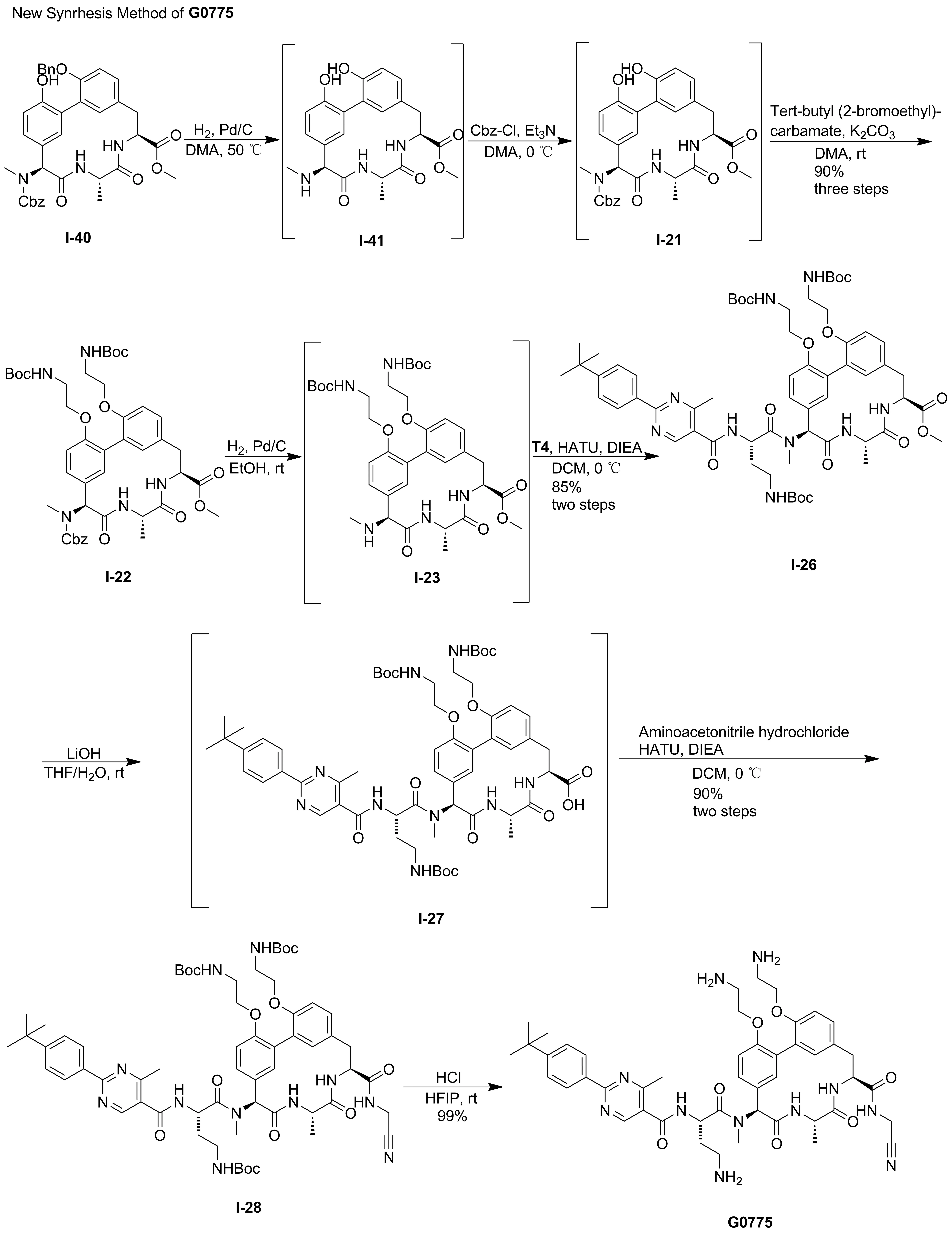
图5 G0775新的合成路线
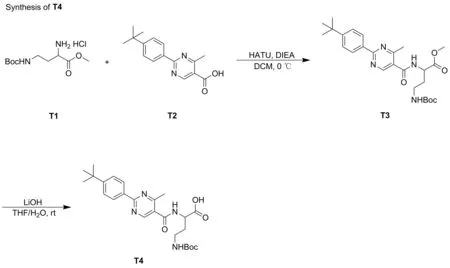
图6 片段T4的合成路线
1 实验部分
1.1 仪器与试剂
Bruker-400/600 MHz型核磁共振仪(TMS为内标,CDCl3和(CD3)2SO为溶剂)、LTQ离子阱质谱仪、Q-TOF四极杆飞行时间质谱仪。
所用溶剂及试剂均为分析纯,硅胶粉200~300目。
1.2 合成
(1) (S)-4-((叔丁氧基羰基)氨基)-2-(2-(4-(叔丁基)苯基)-4-甲基嘧啶-5-甲酰氨基)丁酸甲酯(T3)的合成
向100 mL茄形瓶中依次加入二氯甲烷20 mL和(S)-2-氨基-4-((叔丁氧羰基)氨基)丁酸甲酯盐酸盐2.0 g(7.44 mmol)、 2-(4-(叔丁基)苯基)-4-甲基嘧啶-5-羧酸[11](T2)2.6 g(9.67 mmol)、N,N-二异丙基乙胺3.8 g(29.76 mmol),在0 ℃条件下搅拌反应10 min后加入2-(7-氮杂苯并三氮唑)-N,N,N′,N′-四甲基脲六氟磷酸酯5.6 g(14.88 mmol),继续0 ℃条件下搅拌1 h。反应结束,将反应混合物减压浓缩,并将残余物用乙酸乙酯150 mL溶解,依次用水、10%碳酸钠水溶液和饱和食盐水各150 mL洗涤有机层,有机层经无水硫酸钠干燥,过滤,减压浓缩得粗产物,经柱层析(洗脱剂:石油醚 ∶乙酸乙酯=1 ∶30~1 ∶10,V∶V)纯化,得白色固体T33.4 g,产率95%;1H NMR(400 MHz, Chloroform-d6)δ: 8.87(s, 1H), 8.39(d,J=8.3 Hz, 2H), 7.52(d,J=8.4 Hz, 2H), 4.96~4.86(m, 1H), 3.80(s, 3H), 3.47(s, 1H), 3.18~3.04(m, 1H), 2.77(s, 3H), 2.22~1.99(m, 2H), 1.43(s, 9H), 1.38~1.36(m, 9H);13C NMR(151 MHz, Chloroform-d6)δ: 172.44, 166.58, 166.01, 165.06, 156.31, 155.58, 154.66, 134.27, 128.42(2C, overlap), 125.70(2C, overlap), 79.90, 52.81, 50.17, 36.52, 34.93, 32.63, 31.23(3C, overlap), 28.39(3C, overlap), 23.38; HR-MS(ESI)m/z: Exact mass calcd for C26H36N4O5{[M+H]+}485.2763, found 485.2758。
(2) (S)-4-((叔丁氧基羰基)氨基)-2-(2-(4-(叔丁基)苯基)-4-甲基嘧啶-5-甲酰氨基)丁酸(T4)的合成
向100 mL茄形瓶中依次加入四氢呋喃和水(1 ∶1,V∶V)的混合溶剂20 mL和(S)-4-((叔丁氧基羰基)氨基)-2-(2-(4-(叔丁基)苯基)嘧啶-5-甲酰氨基)丁酸甲酯(T3)2.7 g(5.58 mmol),随后加入氢氧化锂一水合物930 mg(22.32 mmol),混合物室温下搅拌反应1 h,反应结束,向混合物中加入1 M硫酸氢钾水溶液,直到pH达到5,用乙酸乙酯(3×30 mL)萃取,合并有机层,然后用饱和食盐水(2×50 mL)洗涤,有机层经无水硫酸钠干燥,过滤,减压浓缩得到白色固体T42.6 g,产率99%;1H NMR(400 MHz, DMSO-d6)δ: 8.86(d,J=7.6 Hz, 1H), 8.78(s, 1H), 8.40~8.30(m, 2H), 7.60~7.52(m, 2H), 6.91(t,J=5.7 Hz, 1H), 4.44~4.26(m, 1H), 3.16~3.02(m, 2H), 2.62(s, 3H), 2.08~1.91(m, 2H), 1.38(s, 9H), 1.33(s, 9H);13C NMR(126 MHz, DMSO-d6)δ: 175.13, 175.05, 166.93, 166.38, 164.70, 156.87, 155.44, 155.27, 154.71, 133.90, 128.41, 125.81, 125.59, 80.16, 50.80, 34.91, 32.47, 31.21(3C, overlap), 29.71, 28.34(3C, overlap), 23.22; HR-MS(ESI): Exact mass calcd for C25H34N4O5{[M+H]+}471.2603, found 471.2602。
(3)I-22的合成
室温下向100 mL茄形瓶中依次加入N,N-二甲基乙酰胺20 mL、I-402.3 g(3.53 mmol)和10%的钯碳230 mg,在50 ℃和常压条件下进行催化氢化反应6 h后冷却至室温,将反应混合物过滤除去钯碳,滤液无需进一步处理,随后移至0 ℃环境下,依次加入氯甲酸苄酯604 mg(3.53 mmol)和三乙胺535 mg(5.30 mmol),并在0 ℃条件下反应12 h后加入2-溴乙基氨基甲酸叔丁酯5.5 g(24.70 mmol)、碳酸钾7.3 g(52.95 mmol),并在80 ℃条件下反应7 h。反应结束,冷却至室温,将反应混合物用乙酸乙酯200 mL稀释,用水(3×100 mL)和饱和食盐水100 mL洗涤,有机层用无水硫酸钠干燥,过滤,减压浓缩得粗产物,经柱层析(洗脱剂:二氯甲烷 ∶甲醇=1∶30~1∶10,V∶V)纯化,得白色固体2.7 g, 3步产率90%;1H NMR(400 MHz, DMSO-d6)δ: 9.07(t,J=7.5 Hz, 1H), 8.45(dd,J=23.6 Hz, 9.1 Hz, 1H), 7.43~7.27(m, 5H), 7.16(t,J=9.9 Hz, 2H), 7.08(d,J=8.5 Hz, 1H), 7.02(d,J=8.4 Hz, 1H), 6.75~6.58(m, 4H), 5.98(d,J=4.5 Hz, 1H), 4.85(s, 1H), 4.72(q,J=7.0 Hz, 1H), 4.07~3.91(m,J=21.2 Hz, 6.5 Hz, 4H), 3.69(s, 3H), 3.29~3.00(m, 6H), 2.59(d,J=14.3 Hz, 3H), 1.37~1.31(m, 18H), 1.16(dd,J=10.9 Hz, 6.7 Hz, 3H);13C NMR(126 MHz, DMSO-d6)δ: 172.64, 172.50, 170.68, 170.64, 160.04, 156.68, 156.67, 156.11(2C, overlap), 155.95, 155.55, 154.50, 137.53, 137.37, 134.91, 134.75(2C, overlap), 131.19, 130.33, 130.18, 129.54, 128.90, 128.71, 128.66, 128.58, 128.31, 127.99, 127.16, 113.97, 113.59, 78.31, 78.28, 67.59, 67.40, 66.87, 66.46, 64.59, 61.44, 61.41, 52.81, 50.36, 47.98, 32.99, 32.97, 32.06, 28.62, 19.74, 19.61; HR-MS(ESI): Exact mass calcd for C44H58N5O12{[M+H]+}: 848.4073, found 848.4076。
(4)I-26的合成
室温下向100 mL茄形瓶中,依次加入乙醇50 mL和I-222.6 g(3.07 mmol)、 10%的钯碳260 mg和一滴30%的氨水溶液,在室温和常压条件下进行催化氢化反应6 h后将反应混合物过滤除去钯碳,滤液减压浓缩得白色固体,无需进一步处理,直接溶于20 mL的二氯甲烷,依次加入T42.2 g(4.60 mmol)、N,N-二异丙基乙胺1.2 g(9.21 mmol),在0 ℃条件下搅拌10 min后添加2-(7-氮杂苯并三氮唑)-N,N,N′,N′-四甲基脲六氟磷酸酯2.3 g(6.14 mmol),继续在0 ℃条件下搅拌反应1 h。反应结束,将反应混合物减压浓缩,并将残余物用乙酸乙酯100 mL溶解,用水、10%碳酸钠水溶液和饱和食盐水各50 mL洗涤,有机层用无水硫酸钠干燥、过滤,减压浓缩得粗产物,经柱层析(洗脱剂:二氯甲烷 ∶甲醇=1∶30~1∶10,V∶V)纯化,得白色固体I-263.0 g, 2步产率85%;1H NMR(400 MHz, DMSO-d6)δ: 9.12~9.04(m, 1H), 8.98(d,J=7.2 Hz, 1H), 8.79(d,J=4.7 Hz, 1H), 8.45(d,J=8.9 Hz, 1H), 8.35(t,J=6.8 Hz, 2H), 7.57(dd,J=8.9 Hz, 3.5 Hz, 2H), 7.22~7.15(m, 1H), 7.11(s, 2H), 7.02(dd,J=12.3 Hz, 7.0 Hz, 2H), 6.75~6.61(m, 4H), 6.36(s, 1H), 4.92~4.65(m, 3H), 4.07~3.94(m, 4H), 3.69(s, 3H), 3.32(s, 1H), 3.25~2.96(m, 7H), 2.81(s, 2H), 2.65(d,J=5.8 Hz, 3H), 2.07~1.84(m, 2H), 1.34(q,J=2.7 Hz, 2.2 Hz, 32H), 1.27~1.10(m, 7H);13C NMR(126 MHz, DMSO-d6)δ: 172.62, 172.51, 172.44, 170.37, 166.22, 165.53, 163.51, 156.27, 156.06, 155.95(2C, overlap), 155.53, 154.52, 154.47, 134.94, 134.45, 131.27, 129.98, 129.53, 128.97, 128.69, 128.51, 128.31(2C, overlap), 127.60, 126.03(3C, overlap), 114.02, 113.69, 78.30, 78.27, 78.00, 67.62, 59.49, 52.81, 50.43, 67.44, 48.33, 48.04, 37.14, 35.11, 33.04, 32.69, 31.45(4C, overlap), 30.78, 28.62(10C, overlap), 23.19, 19.75; HR-MS(ESI)m/z: Exact mass calcd for C61H84N9O14{[M+H]+}1166.6132, found 1166.6132。
(5)I-28的合成
室温下向50 mL茄形瓶中依次加入四氢呋喃10 mL和I-263.0 g(2.57 mmol),随后移至0 ℃,加入氢氧化锂一水合物432 mg(10.28 mmol)的水溶液10 mL,将混合物在搅拌下逐渐加热至室温,并在室温条件下搅拌反应1 h后加入1 M硫酸氢钾水溶液,直到pH达到5,用乙酸乙酯(3×30 mL)萃取,合并有机层,并用饱和食盐水50 mL洗涤,有机层用无水硫酸钠干燥、过滤,减压浓缩得白色固体,无需进一步处理,直接溶于24 mL二氯甲烷,移至0 ℃,依次加入N,N-二异丙基乙胺996 mg(7.71 mmol),氨基乙腈盐酸盐359 mg(3.86 mmol),随后添加2-(7-氮杂苯并三氮唑)-N,N,N′,N′-四甲基脲六氟磷酸酯2.0 g(5.14 mmol)的N,N-二甲基甲酰胺溶液4 mL,并在0 ℃条件下搅拌反应1 h。反应结束,将反应混合物减压浓缩,并将残余物用乙酸乙酯100 mL溶解,用水、10%碳酸钠水溶液和饱和食盐水各100 mL洗涤,有机层用无水硫酸钠干燥、过滤,减压浓缩得粗产物,经柱层析(洗脱剂:二氯甲烷 ∶甲醇=1 ∶30~1 ∶10,V∶V)纯化,得白色固体I-282.8 g, 2步产率90%;1H NMR(600 MHz, DMSO-d6)δ: 9.01(d,J=7.7 Hz, 1H), 8.97(d,J=7.4 Hz, 1H), 8.79(d,J=6.3 Hz, 1H), 8.73~8.67(m, 2H), 8.39~8.33(m, 3H), 7.59~7.54(m, 2H), 7.22~7.16(m, 1H), 7.11(s, 2H), 7.06~6.96(m, 2H), 6.76~6.60(m, 4H), 6.36(s, 1H), 4.87~4.66(m, 3H), 4.17(t,J=4.8 Hz, 2H), 4.04~3.96(m, 4H), 3.15~2.98(m, 8H), 2.82(s, 2H), 2.65(d,J=5.3 Hz, 3H), 2.03~1.93(m, 2H), 1.34(dd,J=7.2 Hz, 2.4 Hz, 39H);13C NMR(126 MHz, DMSO-d6)δ: 172.75, 172.50, 172.43, 170.26, 166.23, 165.54, 163.52, 156.28, 156.07, 155.96(2C, overlap), 155.53, 154.47(2C, overlap), 135.06, 134.45, 131.22, 130.09, 130.04, 129.57, 129.09, 128.66, 128.58, 128.31(2C, overlap), 127.60, 126.04(2C, overlap), 118.05, 114.05, 113.78, 78.34, 78.28, 78.01, 67.66, 67.49, 59.52, 51.51, 48.35, 48.21, 37.15, 35.11, 33.98, 32.68, 31.45(2C, overlap), 30.78, 28.65(10C, overlap), 27.80, 23.20, 19.85; HR-MS(ESI)m/z: Exact mass calcd for C62H84N11O13{[M+H]+}1190.6244, found 1190.6245。
(6)G0775的合成
室温下向50 mL茄形瓶中依次加入I-28500 mg(0.42 mmol)和六氟异丙醇溶液3 mL、氯化氢的1,4-二氧六环溶液1.6 mL(4 M, 6.30 mmol),并在室温条件下搅拌反应1 h。反应结束,将反应混合物减压浓缩至干燥得白色固体G0775434 mg,产率99%,纯度97%;1H NMR(400 MHz, DMSO-d6)δ: 9.25(dd,J=19.3 Hz, 7.1 Hz, 1H), 8.99(d,J=7.9 Hz, 1H), 8.86(d,J=6.6 Hz, 1H), 8.43(d,J=8.9 Hz, 1H), 8.40~8.33(m, 2H), 8.21(s, 3H), 8.12(d,J=7.2 Hz, 4H), 7.62~7.55(m, 2H), 7.34(s, 1H), 7.21(d,J=5.9 Hz, 3H), 7.16~7.09(m, 2H), 6.77(d,J=4.0 Hz, 2H), 6.35(s, 1H), 5.14(q,J=7.0 Hz, 1H), 4.98(q,J=7.3 Hz, 1H), 4.82~4.66(m, 2H), 4.33~4.16(m, 4H), 3.73~3.64(m, 2H), 3.27(d,J=16.7 Hz, 1H), 3.19~2.91(m, 8H), 2.85(s, 2H), 2.66(d,J=7.6 Hz, 3H), 2.17~1.97(m, 2H), 1.34(d,J=2.9 Hz, 10H), 1.21(q,J=8.0, 7.1 Hz, 3H);13C NMR(126 MHz, DMSO-d6)δ: 172.81, 172.54, 171.46, 170.15, 166.26, 165.66, 163.62, 156.33, 155.15, 154.60, 154.12, 135.33, 134.35, 131.35, 131.00, 130.29, 129.79, 128.70, 128.66, 128.33(2C, overlap), 127.13, 126.08(2C, overlap), 121.73, 118.04, 115.20, 114.98, 67.69, 67.44, 66.09, 65.93, 59.86, 51.39, 48.59, 48.25, 38.62, 38.57, 36.34, 35.13, 34.10, 33.12, 31.44, 29.26, 27.76, 23.34, 19.88; HR-MS(ESI)m/z: Exact mass calcd for C47H60N11O7{[M+H]+}890.4673, found 890.4672。
G0775的文献合成方法是以I-7为起始原料,经22步反应得到,总产率1.40%。该方法最大缺点为线性路线步骤太长,操作繁琐,总产率很低。此外,I-21合成I-22这步反应存在所需的N-Boc-溴乙胺试剂用量很大(10 eq)和反应时间较长(48 h)的问题,最后一步脱Boc保护反应存在需用制备液相纯化和产率较低(47%)的问题。
针对I-22合成存在的问题,本文展开了反应条件优化研究,结果如表1所示:在相对较高的温度下可以明显缩短反应所需的时间并提高产率(Entries 2~4),但温度过高反而会导致产率急剧下降(Entry 5)。降低N-Boc-溴乙胺试剂当量,反应时间有所延长(Entry 6),但可以取得更高的产率。但当继续降低N-Boc-溴乙胺试剂当量时(Entry 7),发现即使延长反应时间反应也不能进行到底,推测可能是N-Boc-溴乙胺不稳定引起。最终确定该步反应的相对较优条件是I-21与N-Boc-溴乙胺物质的量之比为1∶7, 80 ℃反应7 h。
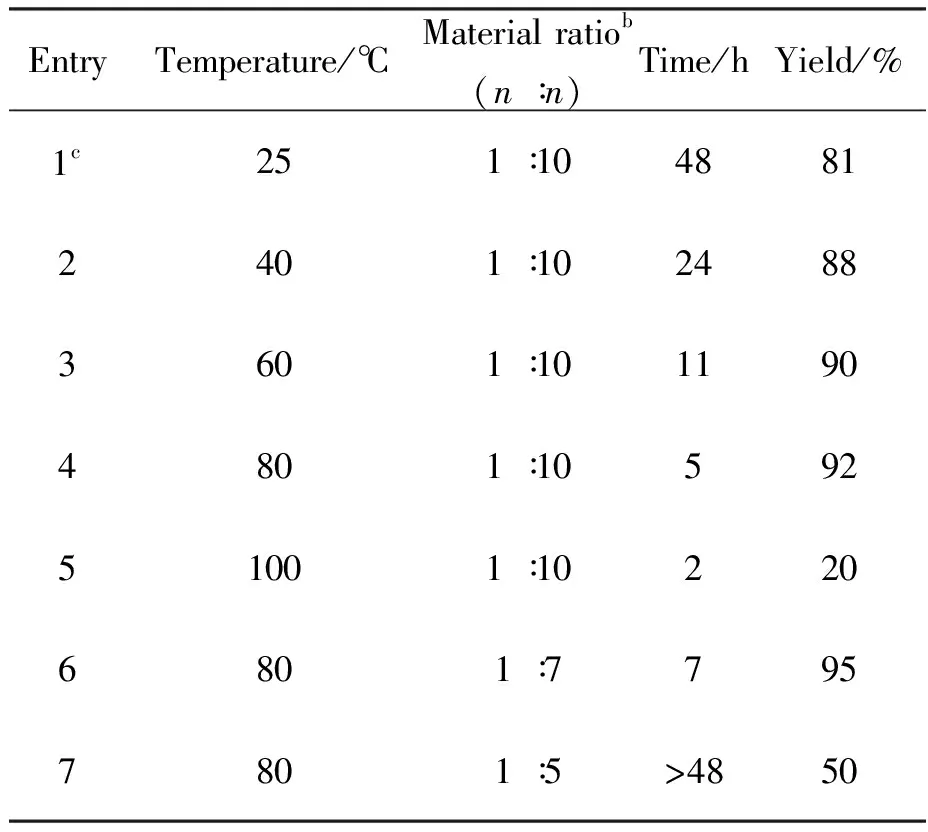
表1 温度和原料配比对于反应时间和反应产率的影响a
最后一步原合成方法需用制备液相纯化且产率较低,探索发现以六氟异丙醇做溶剂,使用氯化氢的1,4-二氧六环溶剂脱保护可以以极高的纯度(97%)和极高的产率(99%)得到G0775盐酸盐,避免了制备液相纯化,产率得到明显提高。
G0775原合成路线步骤长和总产率低的缺点限制了其实用性,新路线则是以I-7为起始原料,仅需15步反应得目标产物G0775,总产率高达23.73%;从关键大环中间体I-40到目标产物G0775,仅需8步,产率为68.16%。本文还巧妙地利用“一锅法”,中间体I-41和I-21不经分离纯化、I-23和I-27仅需简单后处理可直接用于下一步,使得从I-40到G0775共8步反应仅需3步纯化操作即可得终产物,很大程度上减少了操作工序。
本文通过对G0775原合成路线的改善,按照参考文献方法合成中间体甲基(4S,7S,10S)-26-苄氧基-10-((((苄氧基)羰基)甲基)氨基)-16-羟基-7-甲基-6,9-二氧代-5,8-二氮杂-1,2(1,3)-二苯环癸烷-4-羧酸甲酯(I-40),并设计以I-40为起始原料,经过催化氢化、亲核取代、水解和缩合,最后经氯化氢脱保护等共8步反应得到目标产物G0775,总产率为68.16%,纯度97%。相对原合成路线,新路线反应步骤明显缩短,操作更简便,总产率大幅度提高,为该类分子的合成研究提供有益参考。
猜你喜欢
今日农业(2022年13期)2022-11-10
今日农业(2021年15期)2021-11-26
云南化工(2021年2期)2021-05-06
今日农业(2020年17期)2020-10-27
铜仁学院学报(2018年6期)2018-07-05
中国中医药信息杂志(2016年3期)2016-03-02
中国洗涤用品工业(2016年2期)2016-02-28
中国塑料(2015年2期)2015-10-14
云南中医学院学报(2015年2期)2015-07-31
结核与肺部疾病杂志(2015年2期)2015-07-18
