
干法后处理熔盐电解精炼过程数学模型研究
2024-01-22 05:39:24林如山李康祎钟振亚钱宾杰唐洪彬
原子能科学技术 2024年1期
王 赛,林如山,李康祎,钟振亚,钱宾杰,张 磊,唐洪彬
(中国原子能科学研究院 放射化学研究所,北京 102413)
干法后处理是闭式循环快堆核能系统的关键环节[1-2],是高燃耗、高钚含量、强放射性快堆乏燃料后处理的现实技术选择。熔盐电解干法后处理技术具有介质耐辐照、临界风险低、工艺流程短、废物量小等特点,适应性更高、处理对象更广,是最有应用前景的干法后处理技术[3-4]。美俄均已掌握快堆乏燃料干法后处理技术,并分别基于本国燃料循环策略建立了经工程规模热验证的干法后处理流程[5-6]。近年来,我国也已围绕快堆闭式燃料循环,开展了熔盐电解干法后处理技术研究,并取得了较大进步[7-8]。
熔盐电解精炼是乏燃料干法后处理的核心工艺单元,其反应过程复杂、腐蚀工况严苛、高温过程不可直接观察,同时涉及多相界面的流动及传质问题[9]。通过理论模拟和计算分析,研究熔盐电解精炼反应器内的物理及化学变化规律,可为电解精炼工艺优化和设备设计提供参考依据。国外研究人员已利用先进计算机技术,开展了大量研究。如Hoover等[10-11]基于化学动力学、化学热力学和物质传递模型建立了Mark-Ⅳ电解精炼装置的模型,模拟了铀钚锆三种元素在阳极的溶解情况,但该模型不能正确使用过电位。Zhang[12]基于扩散控制假设开发了一种电解精炼过程动力学模型,并用美国阿贡国家实验室(ANL)的实验室数据验证了从液态镉阳极到固体阴极物料变化数据的准确性,该模型考虑了液态电极的体积变化使模拟过程更加精确。Yos等[13]基于扩散层模型和Bulter-Volmer公式建立了电解精炼过程动力学模型,模拟所得物料变化与ANL的实验数据拟合良好,并进一步研究了模型结果对不同输入参数的敏感程度,提出不同输入参数应该考虑不同的扩散层厚度这一结论,该模型缺乏对于固体阳极的适应性研究。国外建立的液态镉阳极的电解精炼模型日趋成熟,对于固体电极的物料变化模拟也有一定进展,但基于物质传递原理对于固体电极的电极电势以及分电流的模拟报道较少。
本文基于电化学热力学及物质传递公式建立乏燃料熔盐电解精炼过程的数学模型,开展铀钚锆三元合金燃料中关键元素的电极电势、分电流及物料分布计算,并采用有限差分法对物料分布变化方程进行离散,通过文献实验数据对建立的数学模型进行准确性验证。
1 数学模型构建
乏燃料电解精炼的电化学行为主要包括乏燃料在阳极溶解、熔盐电解质中的物质传递以及阴极沉积过程,金属乏燃料电解精炼过程原理如图1所示。在实际工程应用中,通过控制电极电位使贵金属裂片产物和Zr保持不溶,形成阳极泥从阳极篮掉落进入Cd相。本文为探究电解精炼规律,在铀钚溶解后继续考虑Zr的溶解。其中涉及到的电化学知识主要包括电池热力学、电极反应动力学以及由迁移和扩散引起的物质传递行为[14],并在此基础上建立数学模型。
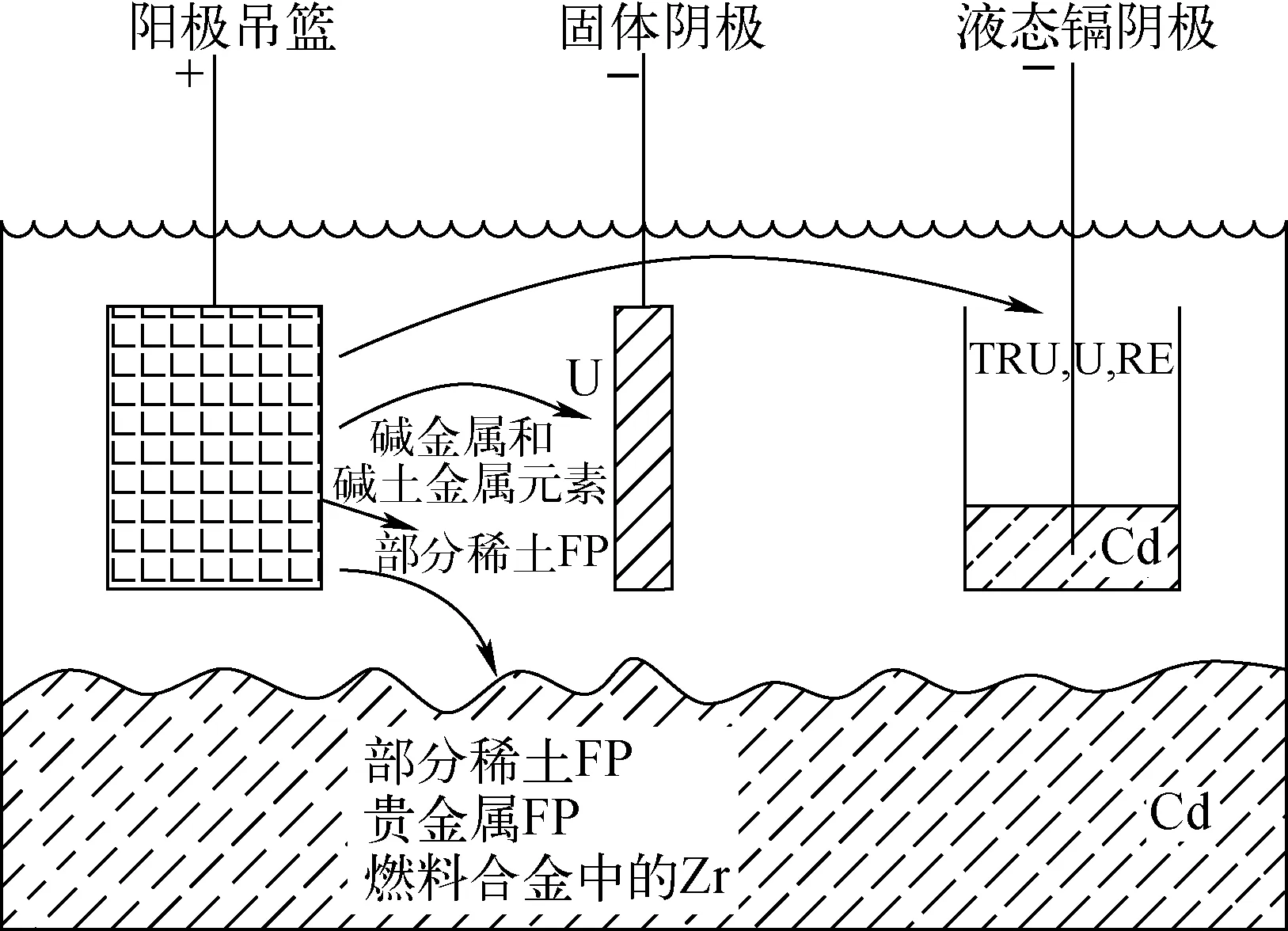
图1 金属乏燃料电解精炼过程原理示意图Fig.1 Schematic diagram of electrorefining process principle of metallic spent fuel
1.1 电化学热力学
以U3+为例,其在阳极发生的反应为:

(1)
根据热力学可逆性,通过Nernst方程可以得到电极电势Eeq和电极过程参与者浓度之间的关系:
(2)
其中:E0为标准电势,V;R为气体常数,取8.314 J/(mol·K);T为开尔文温度,K;F为法拉第常数,取96 485 C/mol;aO和aR分别为电解质中氧化物(O)和还原物(R)的活度。活度与电极状态有关,电极电势因组分不同有不同的表现形式,本文考虑的两种电极均为固体电极。假设元素在固体阴极的活度为1,则电极上的Nernst方程可转化为:
(3)
参与阳极反应的各元素的界面电势差相同,等于阳极电势;参与阴极反应的各元素同理。控制方程可表示为:
Ea=Ei,a=Ej,a=…=En,a
(4)
Ec=Ei,c=Ej,c=…=En,c
(5)
其中:下标a和c分别表示阳极和阴极;i、j、n分别代表参与电极反应的元素。
1.2 物质传递及电流
溶液中的物质传递表现形式为扩散、迁移及对流,假设在电极附近电活性物质的传递全部为扩散形式。根据Fick定律,熔盐电解质与电极界面处元素的摩尔通量(fi)可表示为:
(6)
其中:N为熔盐/电极界面法线;Di为元素i的扩散系数,cm2/s;A为界面面积,cm2;ci为元素浓度,mol/cm3。
假设电极附近扩散层中离子仅在法线方向进行扩散传质。基于量纲分析法,用传质系数(Ki,cm/s)替代式(6)中扩散系数与法线位置偏微分离散形式之商,则在熔盐和固体电极的界面处Fick定律可简化为:
(7)
(8)
基于熔盐中的物质传递,带电荷的元素离子在阳极和阴极产生的分电流为:
(9)
(10)
控制方程为阳极和阴极处的总电流等于各元素分电流之和,阴极电流和阳极电流相等。其表达式为:
(11)
1.3 物料变化方程
本体溶液中元素摩尔浓度随时间变化的微分方程为:
(12)
其中,Vms为熔盐体积,cm3。物质在阳极溶解和阴极沉积的物质的量的变化为:
(13)
其中,Mi为元素i在电极侧的物质的量,mol。
2 电解精炼过程模拟求解
电解精炼过程数学模型的建立通过计算机编程实现。在程序开始前,首先通过假设各元素在阴极与熔盐界面处的浓度为0 mol/cm3,计算阴极允许最大电流:
(14)
式(14)等式右侧各参数均为已知。若施加的电流大于阴极允许最大电流,将发生预测之外的界面反应,应增大熔盐中元素的初始浓度或降低所施加的电流以满足该限制条件。
判定满足电流限制条件后,熔盐性质、元素性质、电解精炼阴阳极尺寸以及物料阳极初始组成及熔盐中元素初始浓度均可通过已知实验数据或计算[15-19]得到。算法流程如下。
1) 在一个合适的范围内估计某时刻阴阳极电极电势,并基于该电势通过式(1)~(2)、(6)~(10)计算电极表面各元素的浓度以及电极与熔盐界面的分电流,通过电流控制方程反向利用二分法拟合逼近阴阳极电极电势。
2) 通过有限差分法离散物料变化微分方程,得到下一时间节点各元素在熔盐中的浓度及摩尔质量数据。分别采用向后差分法和中心差分法离散方程。
中心差分法离散熔盐中元素浓度变化:
(15)
其中,Δt为时间步长,s。
中心差分法离散阴阳极物质的量变化:
(16)
(17)
模型所使用函数数值之间有间断,如某元素在阳极全部溶解后电流突变为0 A。中心差分法在处理此种函数时,具有易振荡不收敛特性。在模拟过程中,发现无法得到收敛解,电极电势、分电流以及物料变化的振荡解如图2所示。
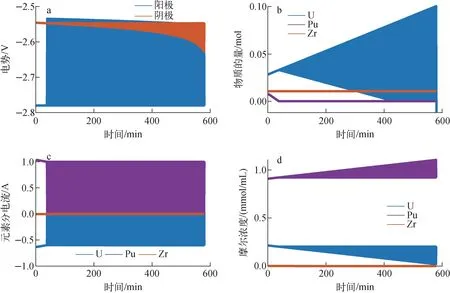
a——电极电势;b——阳极元素物料变化;c——阳极元素分电流;d——熔盐物料变化图2 中心差分法离散物料变化方程的振荡解Fig.2 Oscillation solution of discrete material change equation by central difference method
向后差分法精度为一阶精度,其离散方程如下。
向后差分法离散熔盐中元素浓度变化:
(18)
同样的方法离散阴阳极物质的量变化:
(19)
(20)
3) 时间步长增加1个单位长度,计算传质系数与阳极面积在当下时刻的值。传质系数与阳极面积随电解精炼过程均有不同程度减小,传质系数随时间的变化采用线性函数、阳极面积采用指数函数拟合[11]。返回步骤1。
4) 达到设置的反应时间时,停止程序,并输出阴阳极电极电势、分电流及元素的物质的量在电极侧和熔盐中随时间变化的数据。
程序操作流程如图3所示。
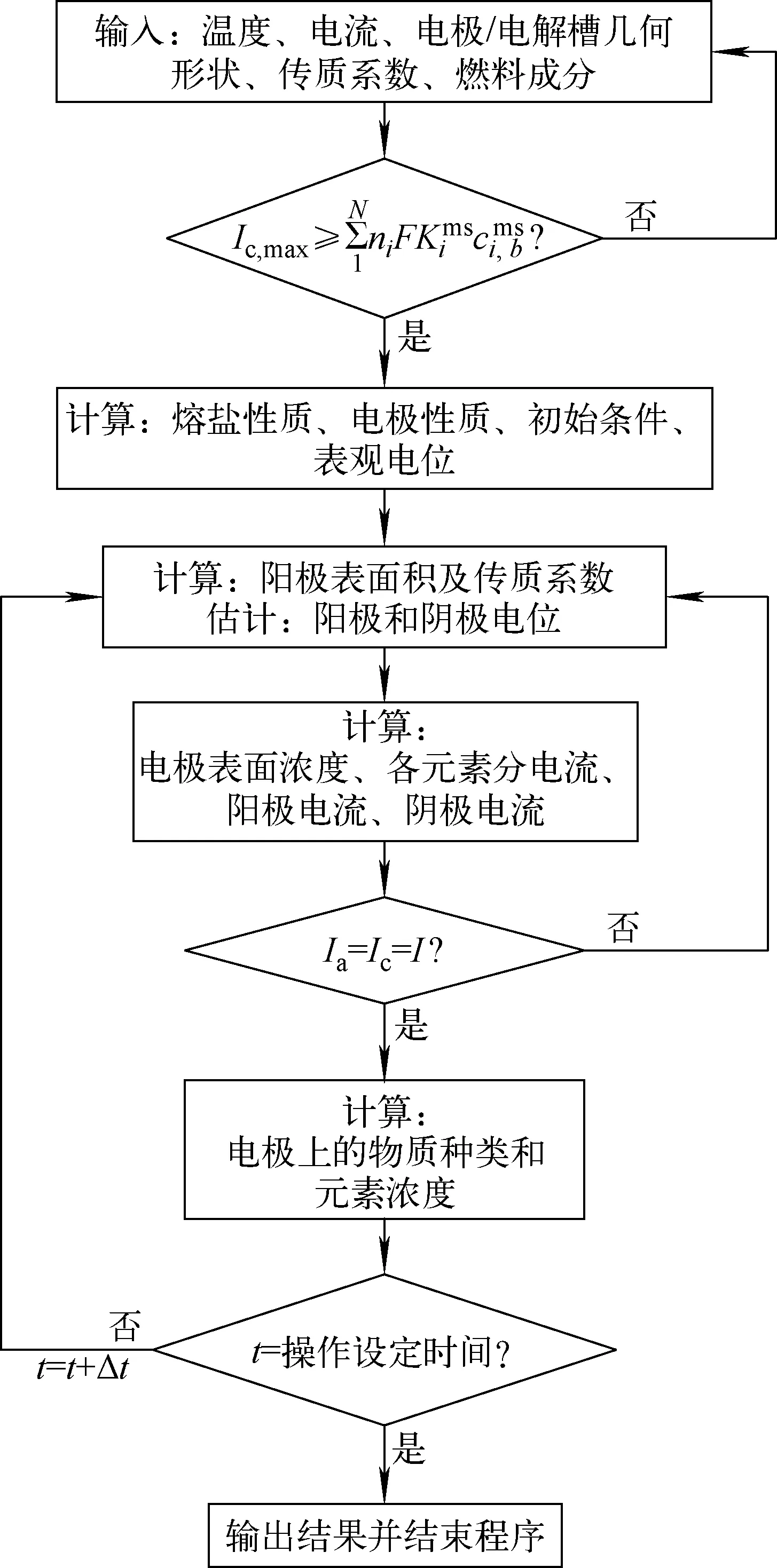
图3 电解精炼程序流程图Fig.3 Program flowchart of electrorefining process
3 结果与讨论
3.1 电解精炼模型结果与验证
本文采用日本中央电力研究所(CRIEPI)的实验研究数据[20]来验证模型的准确性。电解精炼器阳极采用9.6 g三元合金U-20%Pu-10%Zr(质量分数);阴极采用固体不锈钢阴极。容器中放入1 kg熔融电解质,其中U、Pu、Zr的初始质量分数为0.73%、3.1%、0.003%。在753 K下,采用恒电流0.4 A模拟电解精炼过程。其余输入参数与实验条件保持一致。所需物性参数根据文献[15-19]或计算得到,如表1所列,其中各元素标准电势以Cl2/Cl-为参比电极。
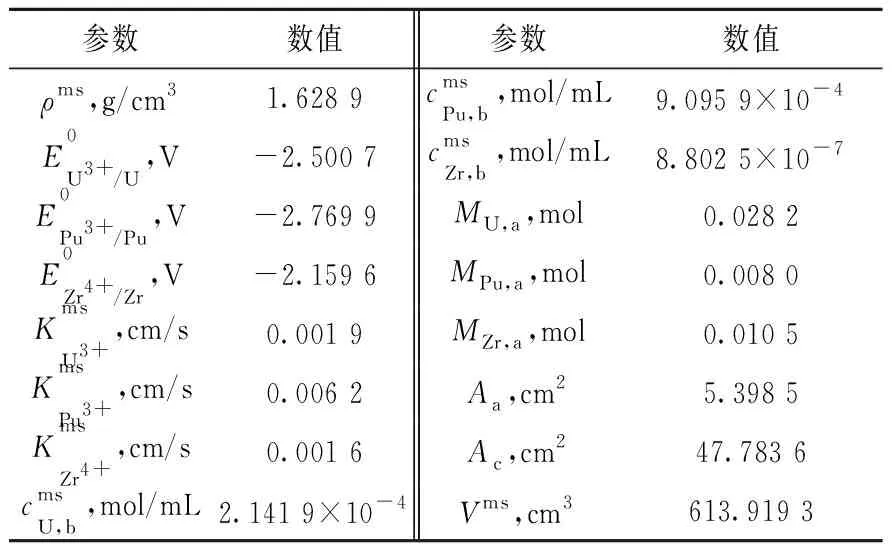
表1 753 K下U、Pu、Zr元素的物性参数Table 1 Physical property parameters of U, Pu and Zr at 753 K
模拟数据与实验数据的对比如图4所示。图4a模拟数据显示阳极电势在30 min附近发生突变,之后呈稳定趋势。模拟数据较实验数据低,这是由于忽略了实验过程存在的电池电阻。由图4b可见,在电解初期,U元素浓度下降,Pu元素浓度升高,之后二者浓度几乎保持不变。整个过程中Zr元素在熔盐中的摩尔浓度始终较低。与实验数据对比可发现,U元素摩尔浓度较真实值高,Pu元素较真实值低,这是因为熔盐中元素浓度对于标准电极电势非常敏感[9],3%的变化会导致熔盐中U元素的量发生50%的偏差。通过电极电势相同以及阴阳极电流相等控制方程得到U元素阴极分电流,进一步根据物料变化方程得到阴极沉积的元素的物质的量。将同一时刻阴极沉积的U计算值0.027 8 mol与实验值0.028 6 mol对比,计算得到相对误差为2.80%。模型充分考虑了U在阴极沉积的过程机理,具有较好的拟合效果。
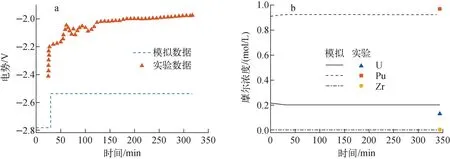
a——阳极电势;b——熔盐中元素物料变化图4 模拟数据与实验数据的对比Fig.4 Comparison of simulated data and experimental data
电流为0.4 A时模拟得到的阴阳极电极电势、各元素分电流及物料随时间变化示于图5。
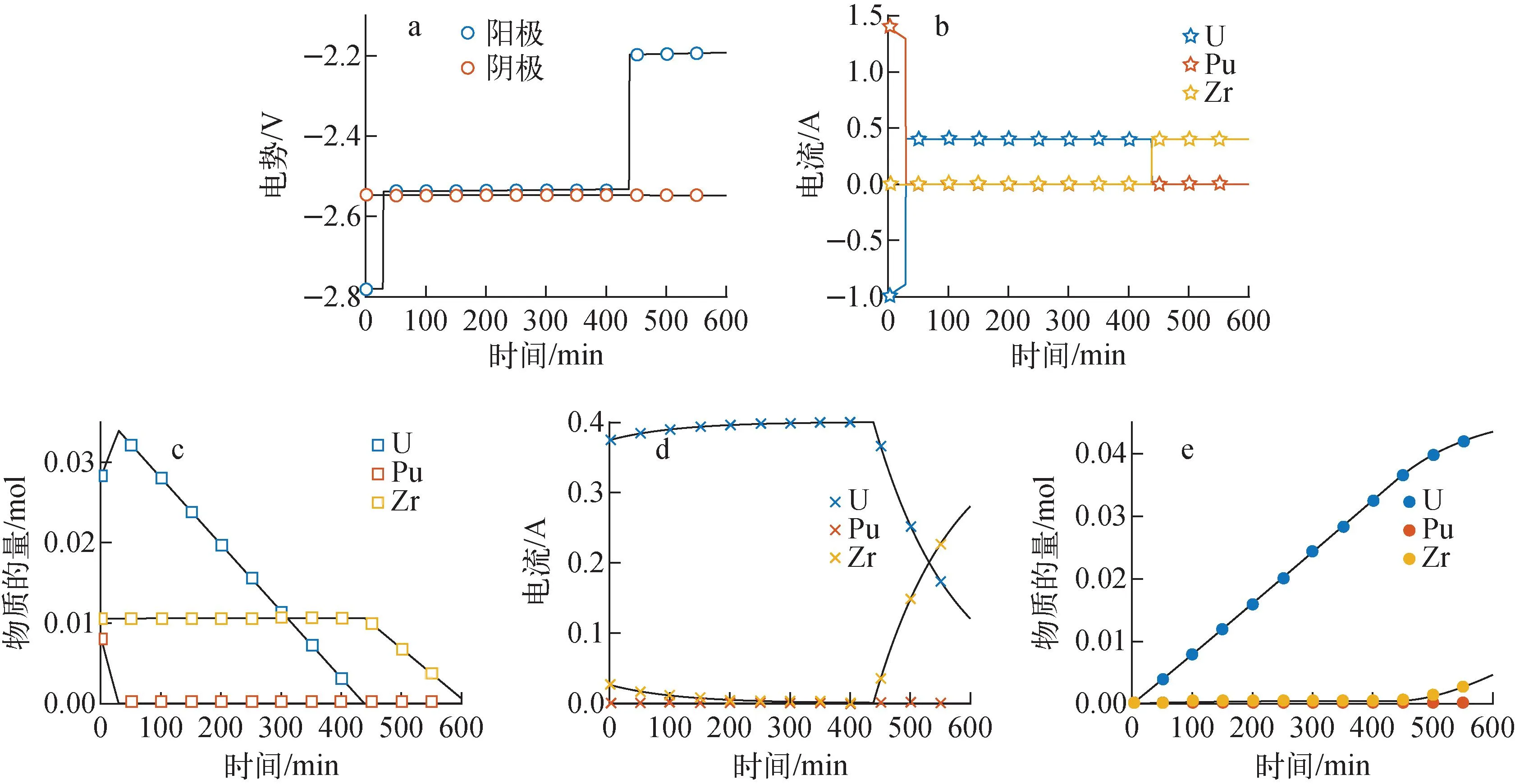
a——电极电势;b——阳极元素分电流;c——阳极元素物料变化;d——阴极元素分电流;e——阴极元素物料变化图5 电流为0.4 A时的模拟结果Fig.5 Simulation results at 0.4 A
由图5a可见,电解精炼器阳极电势存在两次突变。第1次突变表明阳极三元合金的Pu元素完全溶解,U元素开始溶解;第2次突变表明阳极U元素全部溶解,Zr元素开始溶解。电极电势基于Nernst方程计算,模拟结果符合金属活动性顺序表。阴极电势随时间略有减小,初始时刻阴极电势大于阳极电势,之后小于阳极电势。
图5b中,前30 min Pu元素在阳极的分电流从1.4 A线性递减,元素U的分电流从-1 A线性递增,意味着U在阳极原位沉积,该负值电流由置换反应产生;之后Pu元素的分电流突变为0 A,U元素的分电流突变为0.4 A。这一时刻对应阳极电极电势的第1次突变。在439 min时刻,U元素分电流突变为0 A,Zr元素的分电流在此刻从0 A突变为0.4 A。该时刻对应阳极电势的第2次突变。图5c显示,前30 min内,Pu首先在阳极溶解,同时U元素在阳极沉积,质量增加。因为Pu元素除在电流驱动下发生电极反应外,还同时与熔盐中的UCl3发生了置换反应,其化学反应方程式如下:
(21)
Pu在固体电极的表观电位为-2.77 V,远小于U的表观电位-2.5 V,此置换反应可以发生。30 min后,Pu完全溶解,U开始溶解,至439 min时完全溶解,溶解质量与时间呈线性关系。439 min后,惰性金属Zr开始溶解,600 min后在阳极侧完全溶解。
由图5d~e可知,439 min前阴极侧元素分电流大部分由U元素产生。在0~439 min时段,U元素分电流逐渐增大,Zr元素分电流由于浓度降低而不断下降。439 min后,Zr元素由于在阳极溶解浓度升高,分电流逐渐增大,532 min后大于U元素分电流,是电流的主要贡献者。U元素最先在阴极大量沉积,沉积量随时间呈线性变化。同时Zr元素在阴极低速沉积。439 min后,U沉积速率变慢,沉积曲线呈凸函数,Zr元素沉积速率加快。因此为控制金属阴极得到较纯的铀枝晶,应严格控制通电时间,使阳极Zr元素不能溶解留在阳极篮中,同时熔盐中不能出现元素Zr。
3.2 电流强度对电解精炼过程的影响
在电解精炼设备放大过程中,电流强度也将随之增大至百安培级别。将电流强度设置为0.4、0.8、2、4 A,时间截止点为三元合金全部溶解的前一时刻,分析不同电流强度对电解精炼过程的影响,结果示于图6~11。在模拟过程中,阳极合金不断溶解,面积变化采用前文所述指数函数拟合,固体阴极面积不变。

图6 不同电流强度下的阳极电势和阴极电势Fig.6 Anode potential and cathode potential under different current intensities
由图6a可见,随着电流强度的增大,阳极电势整体呈增大趋势,包括Pu溶解时段、U溶解时段以及Zr溶解时段。同时电流增大会使2次电势突变提前,元素溶解的速率加快。因此在放大设备中增大电流有助于阳极乏燃料的溶解。同时不能超出电解精炼过程所允许通过的最大电流限制,若电流大于阴极允许最大电流,可能发生预料之外的化学反应。
由图6b可见,阴极电势整体呈降低趋势。Pu、Zr、U阳极溶解过程阴极电势随时间逐渐线性降低,其中Pu元素斜率最大,U元素斜率最小。
由图7可见,随着电流强度的增大,各元素完全溶解的时间提前。电流扩大2倍后,铀阳极完全溶解时间缩短近1/2。电解精炼初期,U元素在阳极沉积的速率不变,Pu元素在阳极溶解的速率随电流的增大逐渐加快,Zr元素溶解速率与电流强度呈正比。根据模拟结果,在实际电解精炼过程中可以适当提高电流强度,以提高乏燃料在阳极溶解的速率。

图7 不同电流强度下阳极元素物料变化Fig.7 Anode element material change under different current intensities
由图8可见,随着电流的增大,Pu溶解的分电流增大,U在阳极沉积的分电流增大;U溶解和Zr溶解的电流等于输入电流,U溶解结束后,Zr元素溶解电流突变为输入电流。
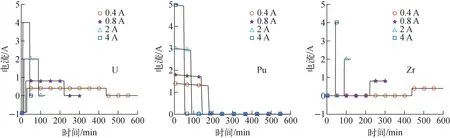
图8 不同电流强度下阳极元素分电流Fig.8 Partial current of anode element under different current intensities
由图9可见,随着时间的增加,U元素大量沉积;阳极Zr元素溶解后,Zr阴极沉积速率增大,电流强度增加,U元素沉积速率加快;Pu元素在阴极沉积的物质的量为0 mol。以上结果表明,增大电流能增大U元素在阴极的沉积速率,因此可在实际应用中增大电流。
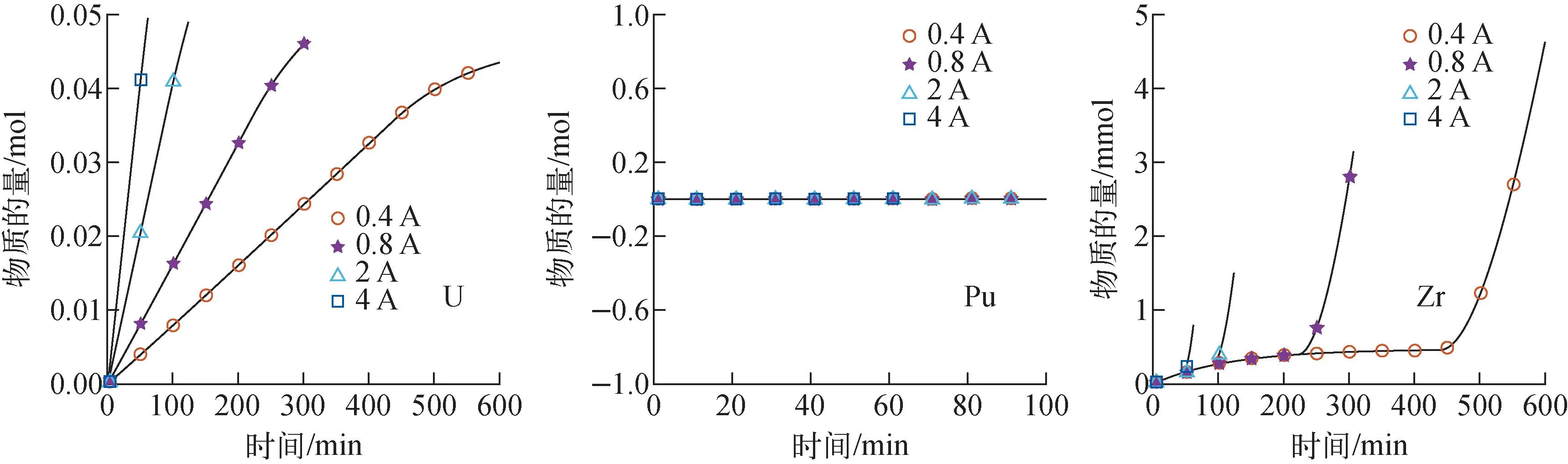
图9 不同电流强度下阴极元素物料变化Fig.9 Cathode element material change under different current intensities
由图10可见,Pu元素的分电流为0 A,U元素分电流占绝大部分。随着Zr元素在阳极的溶解,其分电流逐渐增加。电流增大导致Zr元素的分电流增大速率以及U元素的分电流减小速率加快。电流强度为0.4 A时,在电解精炼后期Zr元素分电流大于U元素分电流,电流强度为0.8、2、4 A时U、Zr元素分电流没有出现交点。
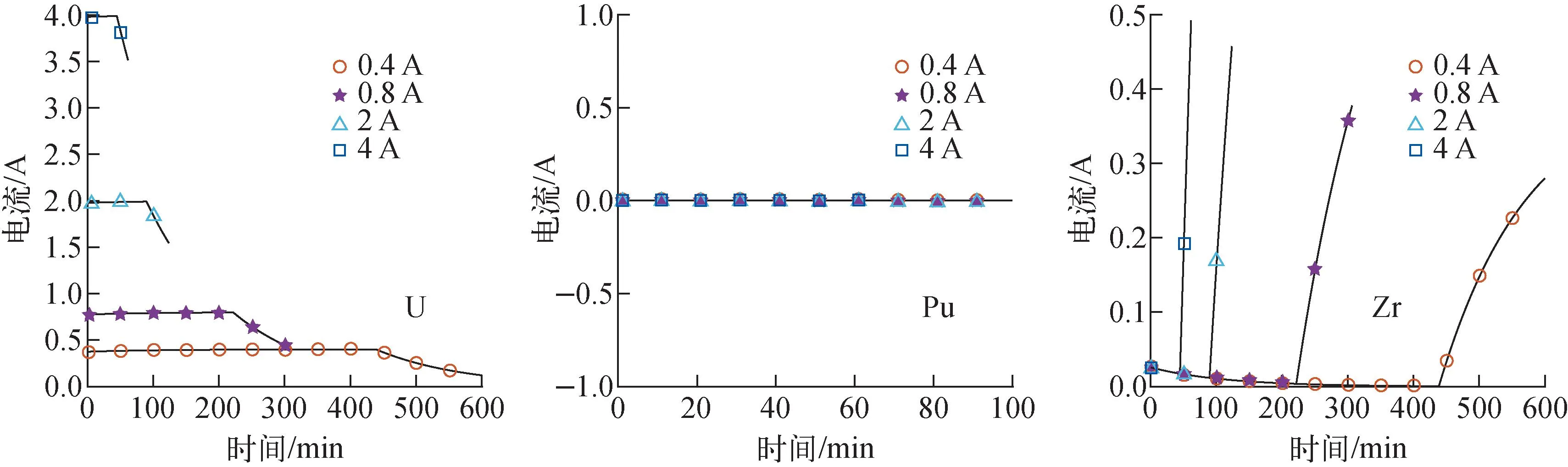
图10 不同电流强度下阴极元素分电流Fig.10 Partial current of cathode element under different current intensities
由图11可见,U元素摩尔浓度在电解精炼初期减小,之后略有增大,在Zr开始溶解后继续降低。随着电流强度的增加,变化速率加快。Pu元素的浓度在不同电流强度下增加至一定量后保持不变,电流越大,增大速率越大,所需时间越短。Zr元素的摩尔浓度先减小后增大,增大速率与电流强度呈正相关。
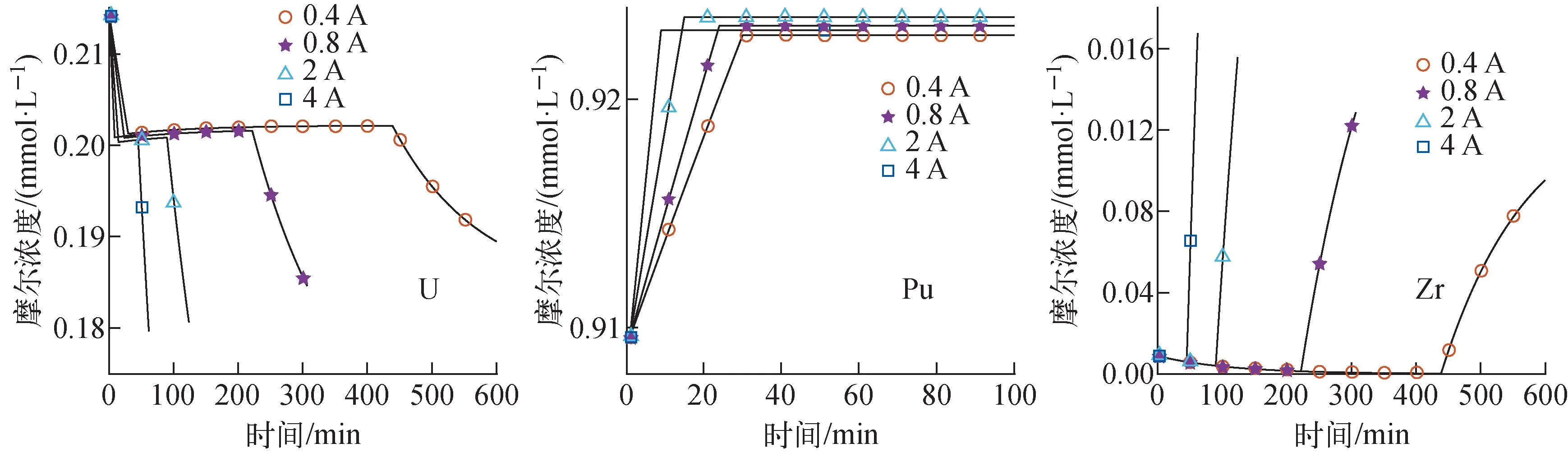
图11 不同电流强度下熔盐中的元素浓度变化Fig.11 Element concentration change in molten salt under different current intensities
4 结论
本文采用有限差分法及二分法建立了铀钚锆三元合金燃料熔盐电解精炼数学模型,计算了电极电势、分电流以及物料分布随时间的变化,通过文献实验数据验证了程序的准确性。
1) 采用向后差分法离散物料变化微分方程建立的电解精炼数学模型可得到收敛结果,避免了中心差分法所建模型计算时的振荡现象。
2) 模拟计算所得阴极沉积铀产品与实验数据的相对误差为2.80%,所建数学模型具有较好的拟合性。
3) 电解精炼过程中,钚铀锆依次在阳极溶解。若锆发生溶解,将在阴极优先沉积,进入铀产品。可根据对铀产品收率和净化的要求,合理控制电极电势。电流强度与电解速率呈正比关系,但不改变钚铀锆的溶解和沉积顺序。
猜你喜欢
Advances in Atmospheric Sciences(2022年6期)2022-04-02 05:29:02
氯碱工业(2021年5期)2021-09-10 07:22:46
中学课程辅导·教师教育(上、下)(2019年22期)2019-12-24 08:58:03
陶瓷学报(2019年6期)2019-10-27 01:18:42
化学教与学(2019年4期)2019-05-14 04:46:40
电子制作(2018年12期)2018-08-01 00:47:46
成都信息工程大学学报(2018年6期)2018-03-21 05:46:14
商情(2017年15期)2017-06-15 11:32:31
材料科学与工程学报(2016年1期)2017-01-15 13:34:02
工业炉(2016年1期)2016-02-27 12:34:11
