
硫化聚丙烯腈凝胶的制备及其锂硫电池性能
2024-01-10 11:32赵永男高海燕
天津工业大学学报 2023年6期
赵永男,李 杨,高海燕,3
(1.天津工业大学天津市先进纤维与储能技术重点实验室,天津 300387;2.天津工业大学材料科学与工程学院,天津 300387;3.南开大学先进能源材料化学教育部重点实验室,天津 300071)
锂硫电池不仅具有高理论容量(1 675 mA·h/g)和高能量密度(2 600 W·h/kg),还具有低成本、环境友好等优点,成为备受关注的新一代电源。然而,由于硫的绝缘性、中间产物多硫化锂的可溶性和电极的体积膨胀所引起的问题,锂硫电池存在活性物质利用率低、循环稳定性差、安全隐患明显等缺陷[1]。大量研究集中于硫载体的结构设计来克服这些缺陷,如微孔[2-3]、介孔[4-5]、分层多孔碳复合材料[6-8]和多孔金属有机框架[9-11]。2003年,Wang 等[12]首次报道了硫化聚丙烯腈(SPAN)复合正极材料,因其高硫利用率、高库仑效率以及良好的循环稳定性而引起了广泛的关注。然而,SPAN 存在影响其实用性的缺陷,如较低的硫含量、较差的高倍率性能以及正极材料的膨胀/收缩问题[13-15]。设计具有适宜孔隙率和高比表面积的多孔结构有助于弥补SPAN的固有缺陷[16]。通常,人们采用工艺复杂的模板法或者原位聚合法制备多孔结构的SPAN 正极材料[17]。本文采用热致相分离法(TIPS)结合化学交联,制备具有三维网络结构的多孔SPAN 凝胶正极材料,提高了SPAN 的锂硫电池性能。
1 实验部分
1.1 试剂与仪器
试剂:聚丙烯腈(PAN,Mav=150 000),天津希恩思科技有限公司产品;二甲基亚砜(DMSO),分析纯,天津市科密欧化学试剂有限公司产品;水合肼(NH2NH2),质量分数80%,天津风船化学试剂有限公司产品;升华硫,纯度为99.5%,上海阿拉丁生化科技股份有限公司产品。
仪器:Hitachi S4800 型扫描电子显微镜,日本日立公司产品;IFS 66V/S 型傅里叶变化红外光谱仪、D8-DISCOVER 型X 射线粉末衍射仪,德国布鲁克公司产品;Autosorb-iQ-C 型物理化学吸附仪,美国康塔公司产品;PGSTAT302N 型Autolab 电化学工作站,瑞士万通有限公司产品;CT-2001A 型LAND 电池测试系统,武汉金诺电子有限公司产品。
1.2 样品的合成
首先将DMSO 和去离子水配制成体积比9 ∶1 的混合液,再将2.0 g PAN 粉末缓慢加入20 mL 混合液中,80 ℃搅拌6 h,至PAN 完全溶解形成溶胶。而后加入0.464 mL 水合肼,继续搅拌2 h,使PAN 分子链完全交联。反应后,将PAN 溶胶迅速转移到培养皿中,于4 ℃静置24 h 使PAN 溶胶发生相分离形成湿凝胶。将PAN 湿凝胶剪成小块并浸泡在去离子水中以除去多余的溶剂和交联剂,同时PAN 湿凝胶固化。将固化的湿凝胶冷冻干燥24 h,得到PAN 干凝胶(记作PANg+N)。
称取适量PANg+N于坩埚中,覆盖过量的升华硫。将坩埚置于管式炉中,在氩气氛围下,以5 ℃/min 升温至155 ℃,保温6 h,再以3 ℃/min 升温至550 ℃,保温5 h,PANg+N和升华硫发生硫化反应生成SPAN 凝胶(记作SPANg+N)。元素分析测定载硫量(质量分数)为32.3%。作为参比样品,还制备了非交联的SPAN 凝胶(SPANg)以及传统SPANp粉末,硫质量分数分别为31.16%和32.24%。
1.3 电化学测试
将正极材料、乙炔黑(AB)、聚偏氟乙烯(PVDF)以8 ∶1 ∶1 的质量比例研磨30 min,然后滴加适量的分散剂N-甲基吡咯烷酮(NMP),继续研磨成均匀的浆料。将浆料涂敷在涂炭铝箔上,60 ℃真空干燥12 h 后,冲裁成直径12 mm 的正极片,其中SPANg+N在极片上的负载量为2.34 mg/cm2。正极片经充分干燥后,以锂片为负极,组装成CR2032 型纽扣电池,电解液为1 mol/L LiPF6/EC+DMC+DEC(1 ∶1 ∶1)。充放电容量均以硫的质量计算。
在1~3 V 的电压范围进行循环伏安(CV)、交流阻抗(EIS)和恒流充放电等测试,其中CV 测试的扫描速率为0.1 mV/s,EIS 测试的频率范围为0.1 ~105Hz。
2 结果与讨论
2.1 结构与物相表征
图1 为PANp、PANg、PANg+N、SPANp、SPANg和SPANg+N样品的SEM 图。

图1 PANp、PANg、PANg+N、SPANp、SPANg 和SPANg+N样品的SEM 图Fig.1 SEM images of PANp,PANg,PANg+N,SPANp,SPANg and SPANg+N
由图1 可见,PANp粉末呈无规则的高度团聚态。凝胶化的PANg呈现规则的多孔结构,孔壁表面光滑,但是孔径较大。交联的PANg+N的结构变化明显,孔径显著变小,孔隙率提高,呈三维网络多孔结构,说明化学交联抑制了相分离过程中PAN 的形变。高温载硫后,SPANp仍然呈高度团聚态,SPANg、SPANg+N则保持了高温处理前的多孔结构,说明PAN 凝胶的结构稳定。
图2 为SPANg+N的TEM 图及EDX 元素面扫图。

图2 SPANg+N 的TEM 形貌图及EDX 元素面扫图Fig.2 TEM morphology of SPANg+N and EDX element mapping
由图2 可见,低倍TEM 图中显示了明显的孔结构,与SEM 结果一致。放大图表明,SPANg+N呈现片层状堆集结构,且有纳米级大孔存在。EDX 元素面扫结果表明,C、N、S 元素在SPANg+N中分布均匀,说明多孔凝胶得到充分硫化。
图3 和表1 分别为PANp、PANg和PANg+N的等温吸脱附曲线和孔结构参数。
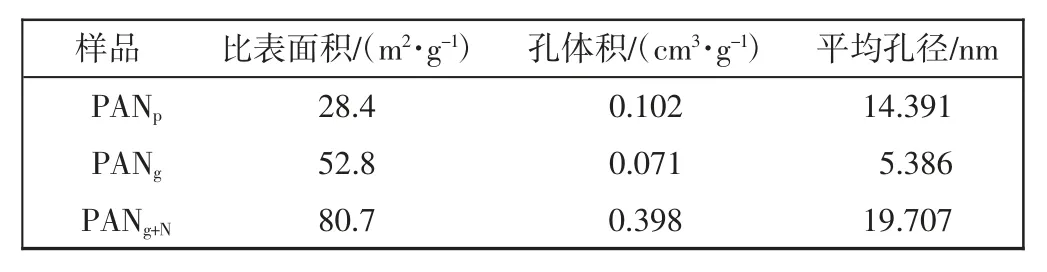
表1 PANp、PANg 和PANg+N 的孔结构参数Tab.1 Hole structure parameters of PANp,PANg and PANg+N

图3 PANp、PANg 和PANg+N 的等温吸脱附曲线(插图为样品的孔分布曲线)Fig.3 Isothermal desorption curves of PANp,PANg and PANg+N(Inset is the pore size distribution curve of the samples)
由图3 可知,PANg+N为Ⅱ型等温吸脱附曲线。P/P0小于0.2 时,氮气吸附量增加缓慢,表明微孔数量很少。当P/P0大于0.8 时,氮气吸附量急剧增加,说明PANg+N中存在大量大孔。滞后环的存在说明PANg+N中存在大量介孔。由表1 可知,PANp、PANg 和PANg+N的比表面积分别为28.4 m2/g、52.8 m2/g、80.7 m2/g,说明凝胶交联处理显著提高了PAN 的孔隙率和比表面积,能够增加单质硫与PAN 基体的接触面积,有利于硫化的进行。
图4 为PANp、PANg和PANg+N的热重分析曲线。
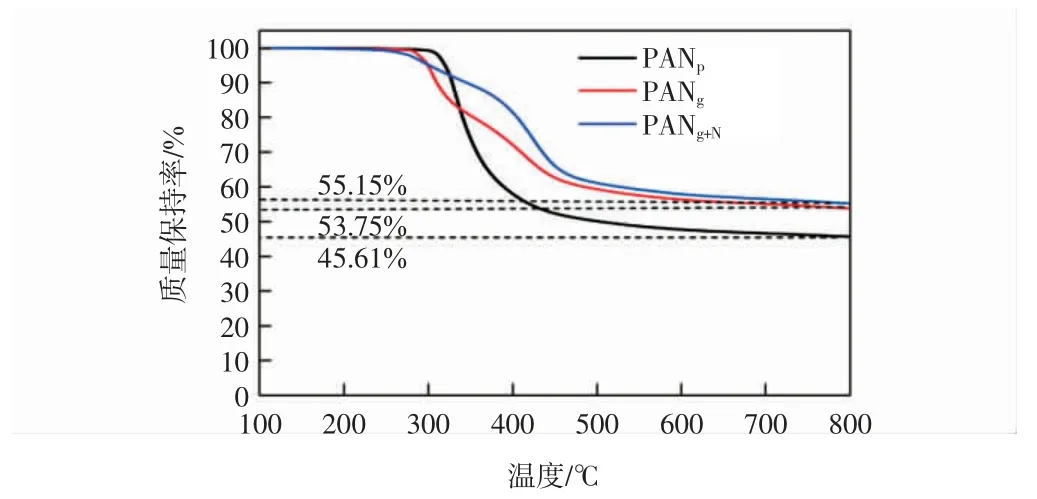
图4 PANp、PANg 和PANg+N 的热重分析曲线Fig.4 TG curves of PANp,PANg and PANg+N
由图4 可知,PANp在280 ℃发生快速质量损失,450 ℃之后,质量损失变得缓慢,至800 ℃趋于平稳,归因于PAN 的热解。PANg在250 ℃存在少量质量损失,主要是材料中溶剂残留。PANg+N在250~450 ℃存在两步质量损失,可能是由于溶剂残留和交联结构断裂所致。3 种材料的最终的碳残量依次为45.61%、53.75%、55.15%,说明凝胶和化学交联有效提高了材料的热稳定性。
PANp、PANg和PANg+N以及SPANp、SPANg和SPANg+N的XRD 衍射图如图5 所示。由图5(a)可见,PANp在17°有一个尖锐的衍射峰,对应PAN 的(100)晶面,在29°的宽峰对应PAN 的(110)晶面,这2 个衍射峰为PAN的特征峰[18],由于PAN 中强极性的氰基使大分子链呈有序堆叠,形成晶相结构[19]。PANg、PANg+N的衍射图和PANp的十分吻合,表明经过凝胶化和水合肼的化学交联没有改变PAN 分子的堆叠方式。由图5(b)可见,硫化后的样品未检测出晶相单质硫和PAN 的衍射,SPANp、SPANg和SPANg+N仅在25°出现宽化的漫衍射峰,说明PAN 和单质硫发生了硫化反应,且硫化聚丙烯腈以无定形的方式存在[20-21]。
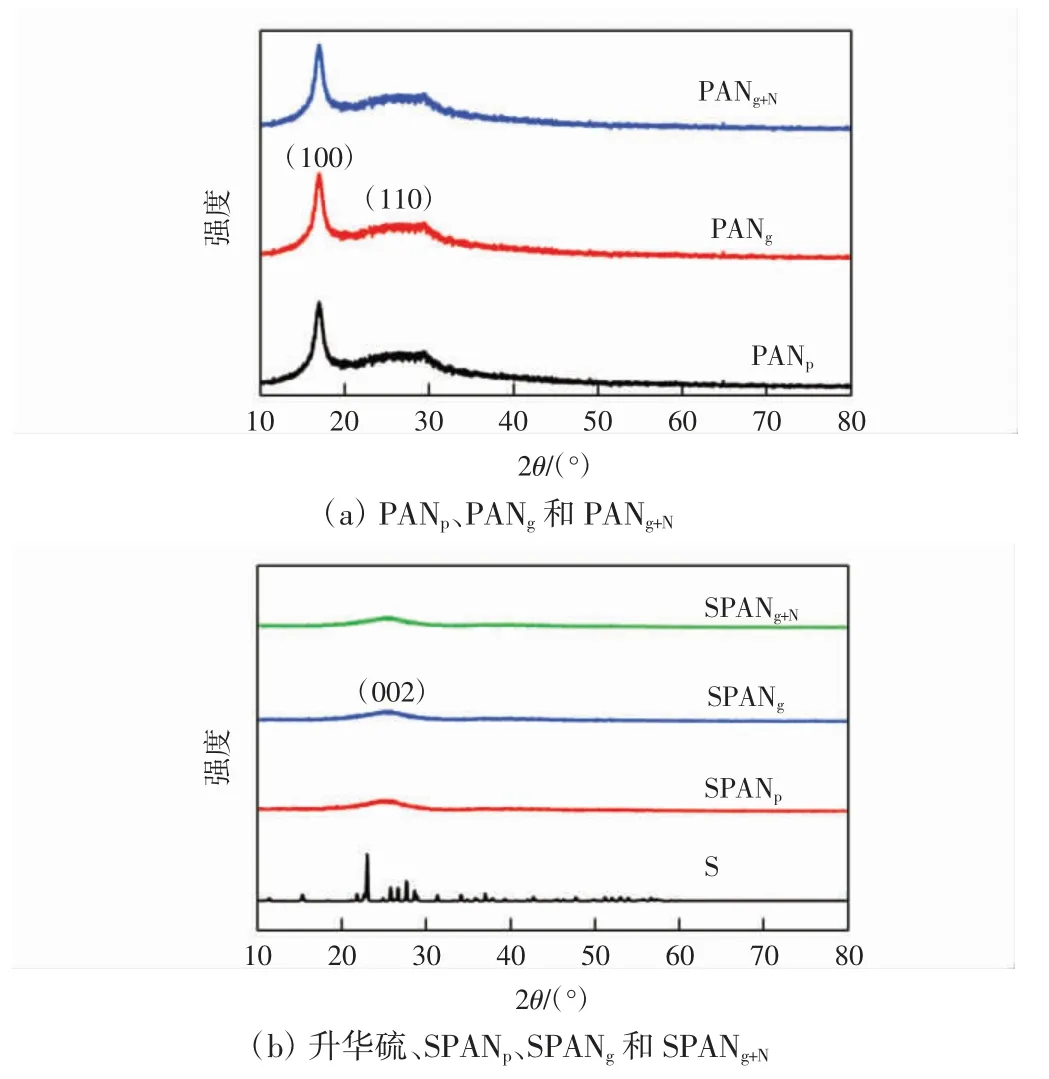
图5 PANp、PANg、PANg+N、升华硫、SPANp、SPANg 和SPANg+N 的XRD 图Fig.5 XRD patterns of PANp,PANg,PANg+N,sublimated sulfur,SPANp,SPANg and SPANg+N
图6 为样品的FTIR 图谱及合成机理图。

图6 样品的FTIR 图谱及SPANg+N 合成机理图Fig.6 FTIR spectra and synthesis mechanism of SPANg+N
由图6(a)可以看出,PAN 在2 243 cm-1的吸收对应的伸缩振动,PANg+N也存在相同的红外吸收。PANg+N在1 652 cm-1和1 697 cm-1分别被检测出基团和—NH—NH—基团的伸缩振动,表明PAN 上的氰基与水合肼相互作用形成—N—N—键,使PAN 分子链被化学交联,如图6(b)所示,证实了水合肼对PAN 分子的交联作用。此外,红外谱图表明,纯PAN凝胶存在氰基间的偶极-偶极作用和分子链间的纠缠效应的物理交联。SPANp、SPANg、SPANg+N的FTIR 图谱均在933 cm-1、669 cm-1、514 cm-1显示了C—S 和S—S的吸收峰[22-23],表明凝胶化和水合肼交联对聚丙烯腈的硫化无影响。
图7 为SPANg+N的XPS 能谱。图7(a)显示了N 的2 种化学态,398.28 eV 的结合能对应的吡啶基氮,399.98 eV 对应C—NH—C 的吡咯氮,而未出现纯PAN 中氰基在399.0 eV 的结合能,表明PAN 在高温硫化过程中发生了脱氢环化。图7(b)显示了硫的3 种化学态,165.18 eV 和163.78 eV 分别对应于硫元素的2P1/2和2P3/2自旋轨道,其中S 2P3/2态比单质硫的结合能(164.0 eV)小,表明SPANg+N中存在C—S 键,而S 2P1/2态对应S—S 键。此外,161.48 eV 的结合能可能源于吸附的高温硫化副产物硫化氢。

图7 SPANg+N 的XPS 能谱Fig.7 XPS spectra of SPANg+N
2.2 电化学性能分析
图8 所示为SPANp和SPANg+N的CV 曲线。

图8 SPANp 和SPANg+N 的CV 曲线Fig.8 CV curves of SPANp and SPANg+N
由图8(a)可见,首圈CV 曲线只出现1 个还原峰,表明充放电过程中没有单质硫的溶解和再沉积过程,只发生了硫和多硫化锂(Li2S/Li2S2)的脱锂/嵌锂的可逆固相反应。首圈还原电位为1.27 V,较低的还原电位可能由于正极表面形成了SEI 膜,是SPAN 正极材料的典型电化学行为。首圈氧化电位为2.33 V,对应放电产物Li2S 在该电位下被氧化。不同于首圈的单还原峰,第2 圈和第3 圈的CV 曲线均出现2 个还原峰,其还原电位分别为1.73 V 和2.12 V,分别对应C—S 键和S—S 键的断裂。与首圈相比,第2 和第3 圈CV 的还原电位发生了明显的偏移,可能是由于放电过程中负载硫的SPAN 发生了体积应力变化。第2 圈和第3 圈的氧化电位均在2.7 V,表明正极材料在充放电过程中的高稳定性。由图8(b)可见,SPANp的还原电位和氧化电位分别为1.06 V 和2.33 V,氧化还原电位差为1.27 V。SPANg+N的还原电位和氧化电位分别为1.27V 和2.28 V,氧化还原电位差仅为1.01 V。较小的电势差表明,SPANg+N在充放电过程中具有更低的极化,说明交联的SPANg+N具有更高的孔隙率,加速了充放电循环过程中离子和电子的传输。
图9 为SPANp和SPANg+N在0.5 C 下的充放电曲线。由图9 可知,SPANp和SPANg+N的首圈充放电均出现低电压的充放电平台,这与上述首圈CV 曲线的结果一致。在第2 圈后的充放电循环中,充放电平台电压增大,且表现出倾斜的平台特征,SPANg+N的放电平台电压显著高于SPANp,且首圈的充放电的电势差小于SPANp,与CV 测试结果吻合。SPANg+N的首圈放电比容量为1 824 mA·h/g,充电比容量为1 395.8 mA·h/g,首圈库仑效率为76.5%,表明了材料具有较好的电化学可逆性。首圈放电比容量高于1 675 mA·h/g 的理论比容量,可能是首圈放电过程中SPAN 的π 共轭吡啶碳参与了嵌锂。
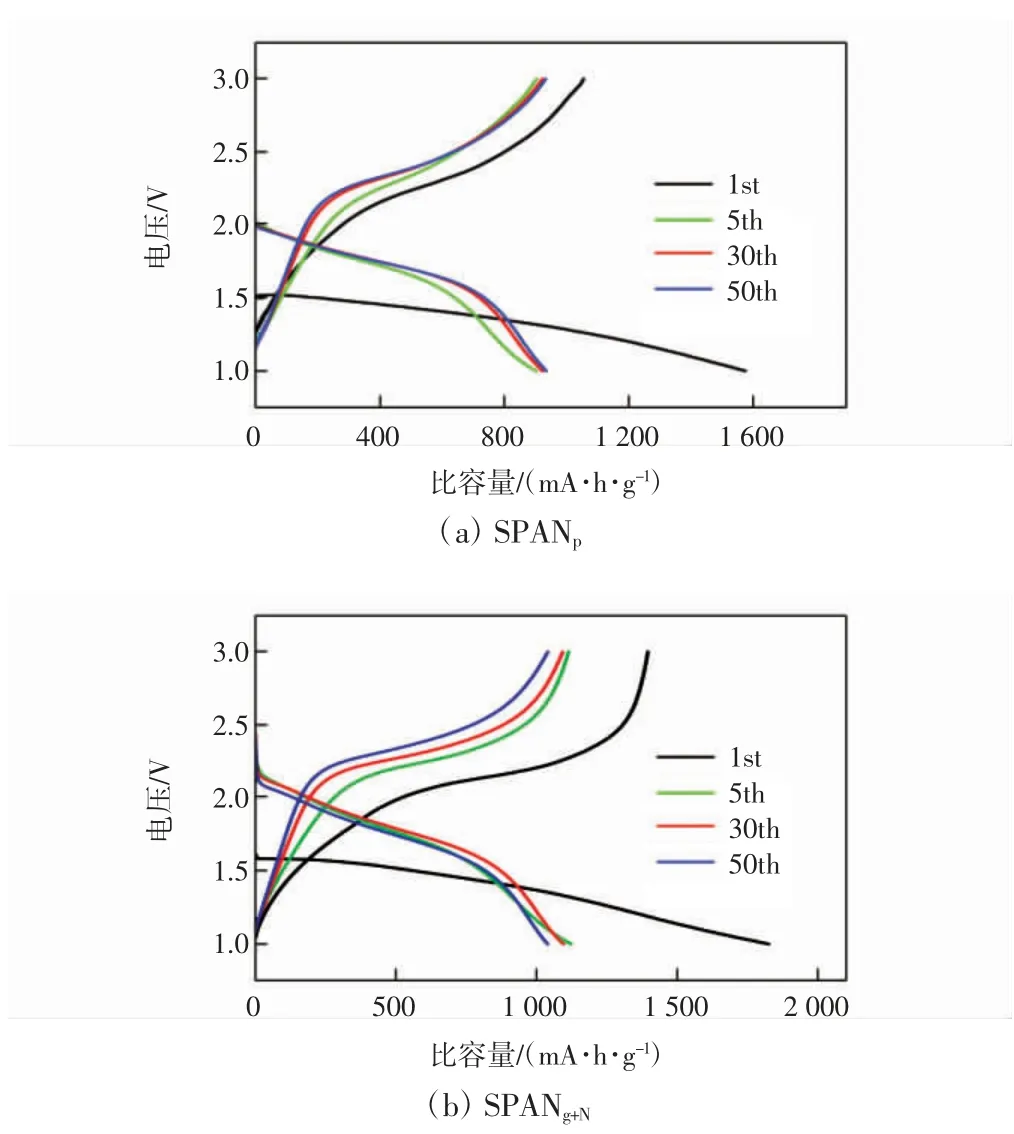
图9 SPANp 和SPANg+N 在0.5 C 下的充放电曲线Fig.9 Charge-discharge curves of SPANp and SPANg+N at 0.5 C
图10 为SPANp、SPANg和SPANg+N在0.5 C 下的充放电循环曲线。

图10 SPANp、SPANg 和SPANg+N 在0.5 C 下的充放电循环曲线Fig.10 Charge-discharge cycling curves of SPANp,SPANg and SPANg+N at 0.5 C
由图10 可知,SPANp、SPANg、SPANg+N的首圈比容量分别为1 175 mA·h/g、1 155.7 mA·h/g 和1 219.8 mA·h/g,循环100 圈后的比容量为966 mA·h/g、995.7 mA·h/g 和1 000.6 mA·h/g。SPANg+N表现出最高的放电比容量,100 圈循环后的容量保持率为82.03%,平均每圈衰减0.19%,说明SPANg+N的三维网络结构提高了材料的容量保持率和循环稳定性。
图11 为SPANp、SPANg和SPANg+N在不同倍率下的充放电曲线。
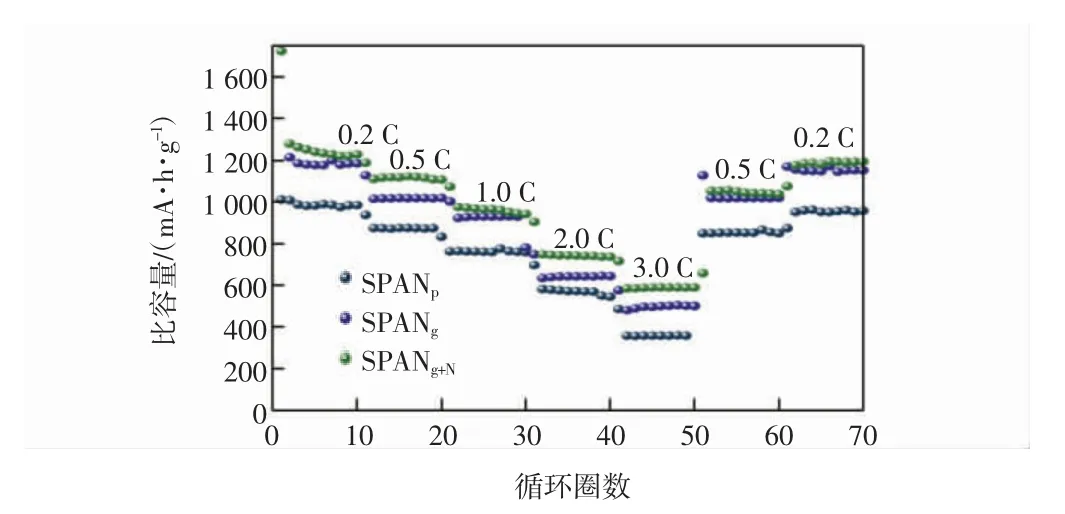
图11 SPANp、SPANg 和SPANg+N 的倍率性能曲线Fig.11 Rate performance curves of SPANp,SPANg and SPANg+N
在图11 中,0.2~3 C 逐渐增大,然后再将电流密度依次减小为0.5 C 和0.2 C,每个倍率充放电10 次,测试电池的倍率性能。由图11 可见,SPANg+N在0.2 C、0.5 C、1 C、2 C、3 C 的首圈比容量分别为1 725.4、1 188.8、1 074.3、905、716.7 mA·h/g。SPANg在不同倍率下的首圈比容量分别为1 784.6、1 128.6、1 003.1、749.3、577.3 mA·h/g。而SPANp在不同倍率下的首圈比容量分别为1 012.7、938.7、763.4、697.7、487 mA·h/g。由此可知,在不同电流密度下,SPANg+N均表现出显著高于SPANp的放电比容量,差值在300 mA·h/g 左右,而SPANg的倍率性能则介于SPANp和SPANg+N之间,说明交联处理有效提高了SPAN 凝胶的倍率性能。此外,电流密度由0.2 C 增大到3 C,SPANg+N的平均放电比容量由1 373.59 mA·h/g 下降到589.7 mA·h/g,容量保持率为42.93%。当电流密度恢复到0.2 C 时,电池的比容量为1 187.88 mA·h/g,容量保持率为86.48%,表明SPANg+N具有良好的电化学可逆性和倍率性能。
SPANp、SPANg+N的电化学阻抗谱如图12 所示。

图12 SPANp 和SPANg+N 的电化学阻抗谱Fig.12 Electrochemical impedance spectra of SPANp and SPANg+N
在图12 中,高频区半圆的直径对应于电荷转移阻抗(Rct),低频区的倾斜直线代表离子扩散系数。可以发现,Rct(SPANg+N)<Rct(SPANp),说明通过凝胶化和化学交联制备的高孔隙率PAN 凝胶的多孔结构显著提高了Li+和电子的传输速率,提高了正极材料的电化学性能。
3 结 论
(1)以热致相分离法制备了聚丙烯腈凝胶,并通过水合肼的化学交联,制备出多孔的交联聚丙烯腈凝胶(SPANg+N)。凝胶化的SPAN 具有高度互联的三维网络结构,化学交联提高了SPAN 凝胶的孔隙率和热稳定性。
(2)高孔隙率的三维网络结构提高了离子和电子迁移速率,改善了SPAN 凝胶的电化学性能。在0.5 C的电流密度下,SPANg+N的首圈比容量达到1 219.8 mA·h/g,循环100 圈后的比容量为1 000.6 mA·h/g,容量保持率为82.03%,平均每圈衰减0.18%。
(3)倍率测试发现,SPANg+N从0.2 C 的电流密度升高到3 C 的电流密度时,比容量从1 373.59 mA·h/g下降到589.7 mA·h/g,容量保持率为42.93%,当重新恢复到0.2 C 时,比容量为1 187.88 mA·h/g,容量保持率可达86.48%。
猜你喜欢
机电产品开发与创新(2023年5期)2023-10-23
氯碱工业(2022年6期)2022-11-21
化工环保(2021年2期)2021-04-25
云南化工(2020年5期)2020-06-12
盐科学与化工(2019年11期)2019-12-04
安徽电子信息职业技术学院学报(2019年2期)2019-04-26
中国特种设备安全(2019年3期)2019-04-22
制造业自动化(2017年2期)2017-03-20
山东工业技术(2016年15期)2016-12-01
中国塑料(2015年6期)2015-11-13
