
芫花素与水溶性磷酸盐柱[6]芳烃包合物的制备、表征及分子模拟研究
2024-01-09 00:45杨云汉杨明坤张郡童杨玉香钏永明杨丽娟
中草药 2024年1期
陶 欣,杨云汉,杨明坤,张郡童,陈 艳,杨 举,杨玉香,钏永明,杨丽娟
云南民族大学化学与环境学院 云南省高校智能超分子化学重点实验室 生物基材料绿色制备技术国家地方联合工程中心,云南 昆明 650500
芫花素又称为5,4′-二羟基-7-甲氧基黄酮,是一种从瑞香科瑞香属植物芫花,唇形科植物刺糙苏、迷迭香和月桂叶、黑桤木、沉香树等众多植物中分离出来的典型的非糖基化类黄酮类化合物[1-3],具有较高的生物活性和低毒性等特点[4]。芫花素具有抗菌[5]、抗炎[6]、抗氧化[7]、抗肿瘤[8-9]、抗疟原虫[10]、自由基清除[11]、免疫调节[4]和抗神经退行性疾病[12]等多种生物药理学作用,近年来被广泛应用于临床上新型药物制剂的研究。但因其性状不稳定,易吸水潮解霉变,需在低温条件下密封干燥保存,溶解性较差[13]。因此亟需找到一种高效低毒的药物载体以改善芫花素在药物制剂研发中受到的限制。
大环化学发展了冠醚[14]、环糊精[15]、杯芳烃[16]、葫芦脲[17]和柱芳烃[18]等5 类经典的大环主体化合物,推动了超分子化学的快速发展。其中柱芳烃作为第5 代大环分子,具有独特的刚性富电子空腔、易于修饰、主客体亲和力强和优异的生物兼容性等特点,使其成为构筑功能有机超分子体系的有利载体[19]。与其他几类经典的大环主体化合物相比,柱芳烃因具有优异的生物兼容性和水溶性[20],被广泛应用于生物医药[21]、功能材料[22]、高效催化剂[23]和环境污染物治理[24]等领域,且成为继环糊精之后一种新型且高效低毒的天然药物载体,能有效改善药物分子的理化性质[25]。
本研究选用水溶性磷酸盐柱[6]芳烃(phosphate salt pillar[6]arene,PP6A)作为药物载体,芫花素为客体分子,采用饱和水溶液法制备芫花素和PP6A的超分子包合物(GK/PP6A),通过紫外-可见吸收光谱法(UV-Vis)、半经验分子轨道法、分子对接、核磁共振波谱(1H-NMR、2D-NMR)、傅里叶变换红外光谱(Fourier transform infrared spectroscopy,FT-IR)、X 射线粉末衍射(X-ray powder diffraction,XRD)、扫描电子显微镜(scanning electron microscope,SEM)和差示扫描量热(differential scanning calorimetry,DSC)等分析方法对芫花素包合前后的理化性质进行研究分析,并推测了主客体可能的包合模式。进一步以包合物的包合率为指标,对GK/PP6A 包合物的制备工艺进行了优化筛选。
1 仪器与试剂
Nova Nanosem-450 型扫描电子显微镜,美国FEI 公司;Bruker D8 Advance A25X 型X 射线粉末衍射仪、Bruker Avance 400 型核磁共振仪,瑞士Bruker 公司;UV-2600i 型紫外分光光度计,日本岛津仪器有限公司;Nicolet IS10 型傅里叶红外光谱仪,美国Thermo Fisher Scientific 公司;STA 449F3型差示扫描量热仪,德国Netzsch 公司;S22-2 型恒温磁力搅拌器,上海司乐仪器有限公司;N-1100DWD 型旋转蒸发仪,云南科仪化玻有限公司;SHZ-(D)III 型循环水式真空泵,巩义市英峪予华仪器厂;FA1004 型电子分析天平,上海天平仪器厂;DZF 型真空干燥箱,上海一恒科学仪器有限公司。
三苯基膦(质量分数99.0%,批号P1475928)、三甲基溴硅烷(质量分数98.0%,批号P1348856)、亚磷酸三乙酯(质量分数98.0%,批号P1656460)购于阿拉丁试剂有限公司;四溴化碳(质量分数97.0%,批号P1412529)、1,4-二(2-羟基乙氧基)(质量分数95.0%,批号F1220202)、多聚甲醛(质量分数96.0%,批号P1322770)、KBr(质量分数99.99%,批号20200401)购于泰坦科技有限公司;三氟化硼乙醚(质量分数46.5%,批号9LWDR4VQ)购于安耐吉化学有限公司;氨水(体积分数25.0%,批号2022041326)购于天津市致远化学试剂有限公司;芫花素(质量分数98.0%,批号HR1168W9)购于陕西辰光生物科技有限公司;氯仿(体积分数99.0%,批号20220301)、丙酮(体积分数99.5%,批号20220301)购于重庆川东化工有限公司。
2 方法与结果
2.1 PP6A 的合成及其溶解度研究
2.1.1 PP6A 的合成 参照文献方法[26]合成PP6A,合成路线如图1 所示,产率98.20%。谱图数据与文献报道一致,1H-NMR (400 MHz,D2O)δ: 6.95 (12H,s), 4.09 (24H, s), 3.84 (12H, s), 2.13 (24H, s);13C-NMR(100 MHz, D2O)δ: 150.1, 128.8, 116.8, 65.4, 29.3,28.3。

图1 PP6A 的合成路线图Fig.1 Synthesis route of PP6A
2.1.2 PP6A 的溶解度研究 用超纯水配制系列质量浓度梯度的PP6A 溶液,并在最大吸收波长(λ=340 nm)处测定其紫外吸收光谱,以PP6A 吸光度(A)对其质量浓度(C)作图,得线性回归方程A=18.511 43C-0.000 641,r=0.999 5,结果表明PP6A在5.991 3~60.137 9 mg/L 线性关系良好。
通过饱和溶液法探究了不同温度下PP6A 的溶解度,分别在25、30、40、50 ℃的温度下制备PP6A的饱和水溶液,并通过PP6A 的紫外吸收标准曲线计算出PP6A 在不同温度下的溶解度,如表1 所示。由实验结果可知,随着温度的升高,PP6A 的溶解度逐渐增大。
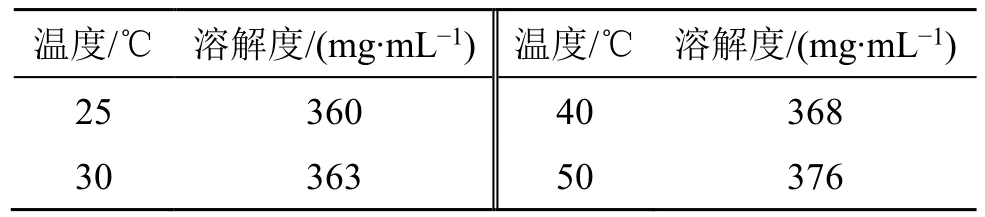
表1 PP6A 在不同温度下的溶解度Table 1 Solubility of PP6A at different temperatures
2.2 GK/PP6A 包合物的制备处方工艺筛选与优化
2.2.1 GK/PP6A 包合物的制备 以PP6A 作为芫花素的药物载体即主体分子,芫花素为客体分子,采用饱和水溶液法制备GK/PP6A 包合物。按照不同物质的量比(PP6A∶芫花素)准确称取定量的PP6A与芫花素;用不同比例的无水甲醇与水作为溶剂,将芫花素溶解于甲醇中超声30 min 使其充分溶解,同时将PP6A 放入圆底烧瓶中加入适量的超纯水将其溶解,并于不同温度下搅拌使其充分溶解,然后将提前准备好的芫花素溶液缓慢加入搅拌一段时间,反应结束后,处理滤液得GK/PP6A 包合物。
2.2.2 芫花素线性关系考察 使用UV-2600i型紫外分光光度计进行紫外-可见光谱测试,研究芫花素的标准曲线时,采用DMSO 与水的混合溶剂(1∶1),并在最大吸收波长(λ=340 nm)处测定紫外吸收光谱。配制质量浓度为10 mmol/L 的芫花素溶液,然后依次取2、4、6、8、10、12、14、16、18 μL 的芫花素溶液,分别稀释到3 000 μL 的溶液(DMSO-水1∶1)中,并在最大吸收波长处(λ=340 nm)测定紫外吸收光谱,以A值对质量浓度(C)作图,得线性回归方程为A=16.572 62C-0.001 35,r=0.999 6,结果表明芫花素在1.904 5~17.169 3 mg/L线性关系良好。
2.2.3 单因素实验考察GK/PP6A 包合物的制备处方工艺
(1)PP6A 与芫花素物质的量比:固定包合温度为30 ℃,包合时间为8 h,甲醇与水的体积比为1∶8,分别在PP6A 与芫花素的物质的量比为1∶1、1∶2、1∶3、1∶4 的条件下制备包合物,通过包合率考察不同物质的量比对包合工艺的影响。结果包合率分别为30.13%、46.37%、44.26%、37.62%,结果表明,当PP6A 与芫花素的物质的量比为1∶2 时,包合率最大,故选择PP6A 与芫花素物质的量比的研究范围为1∶1~1∶3。
(2)包合温度:固定PP6A 与芫花素的物质的量比为1∶2,包合时间为8 h,甲醇与水的体积比为1∶8,分别在包合温度为20、30、40、50 ℃的条件下进行包合物的制备,通过包合率考察不同物质的量比对包合工艺的影响。结果包合率分别为38.17%、45.63%、45.11%、41.98%,结果表明,当包合温度为30 ℃时,包合率最大,故选择包合温度的研究范围为30~50 ℃。
(3)包合时间:固定PP6A 与芫花素的物质的量比为1∶2,包合温度为30 ℃,甲醇与水的体积比为1∶8,分别在包合时间为6、8、10、12 h 的条件下制备包合物,通过包合率考察不同物质的量比对包合工艺的影响。结果包合率分别为32.07%、46.48%、45.01%、43.21%,结果表明,当包合时间为8 h 时,包合率最大,故选择包合时间的研究范围为6~10 h。
(4)甲醇与水的体积比:固定PP6A 与芫花素的物质的量为1∶2,包合温度为30 ℃,包合时间为8 h,分别在甲醇与水的体积比为1∶4、1∶6、1∶8、1∶10 的条件下制备包合物,通过包合率考察不同物质的量比对包合工艺的影响。结果包封率分别为29.76%、37.35%、48.59%、43.78%,结果表明,当包合溶剂体积比为1∶8 时,包合率最大,故选择甲醇与水体积比的研究范围为1∶6~1∶10。
2.2.4 正交试验优化GK/PP6A 包合物的制备处方工艺 通过单因素试验得出的各试验因素水平,以包合率[27]为评价指标设计正交试验,以PP6A 与芫花素的物质的量比(A)、包合温度(B)、包合时间(C)和甲醇与水的体积比(D)为因素,每个因素3个水平,选用L9(34) 设计正交试验,试验设计与结果见表2。通过正交试验直观分析结果表明,4 种因素对包合率的影响顺序为D>A>C>B,即甲醇与水的体积比>PP6A 与芫花素的物质的量比>包合时间>包合温度。由测试结果可知,最优制备工艺为A1B3C2D3,当PP6A 与芫花素的物质的量比为1∶1,包合温度为50 ℃,包合时间8 h,甲醇与超纯水体积比为1∶8 时包合率最高,为89.93%。

表2 L9(34)正交试验设计与结果Table 2 Design and results of L9(34) orthogonal test
从表3 可知,因素A 和D 在不同水平间存在差异(P<0.05),为GK/PP6A 包合物的制备工艺中较为重要的影响因素,B 和C 因素在不同水平间不存在显著性差异,为次要影响因素。

表3 方差分析Table 3 Analysis of variance
2.2.5 GK/PP6A 包合物的制备处方工艺确定 采用“2.2.3”项中的优化方法制备GK/PP6A 包合物,准确称取PP6A(14.2 mg,M=2 243.980 g/mol,6.30 μmol),芫花素(1.8 mg,M=284.268 g/mol,6.30 μmol),将芫花素溶解于1.5 mL 的甲醇中超声30 min 充分溶解,同时将PP6A 放入25 mL 圆底烧瓶中,加入12 mL 的超纯水使其充分溶解,然后将提前准备好的芫花素溶液缓慢加入烧瓶中,50 ℃搅拌8 h。反应结束后旋转蒸发除去溶剂,并向圆底烧瓶中加入适量超纯水超声溶解,用0.45 μm 的醋酸纤维滤头滤除滤液中未进入柱芳烃空腔的,沉淀于溶液中的芫花素,再次旋转蒸发除去溶剂,真空干燥,得GK/PP6A 包合物。
2.3 GK/PP6A 包合物的表征
2.3.1 FT-IR 分析 通过Nicolet IS10 傅里叶变换红外光谱仪测试芫花素、PP6A 和GK/PP6A 的红外光谱信息。采用压片法将样品与溴化钾(样品-KBr 1∶100)研磨制成薄片测定红外吸收光谱。PP6A、芫花素和GK/PP6A 包合物的FT-IR 如图2 所示,从主体分子PP6A 的红外吸收光谱图中可以看出1 217、1 144 cm−1处为PP6A 分子上P=O 的伸缩振动吸收峰,797、702、546 cm−1处为PP6A 苯环上C-H 的弯曲振动吸收峰。芫花素的红外吸收光谱显示了其在3 269 cm−1处具有羟基-OH 的伸缩振动峰,2 935 cm−1处为芫花素21号原子处的-CH3的伸缩振动峰,1 669 cm−1处为羰基的伸缩振动,1 499 cm−1处为苯环上的C=C 骨架伸缩振动,1 119 cm−1处为C-OC 的弯曲振动。当芫花素与PP6A 形成包合物后,芫花素在3 269 cm−1处的-OH 伸缩振动峰消失,2 935、1 669、1 499、1 119 cm−1处的红外吸收特征峰均明显减弱,而1 217、1 144、797 cm−1处的PP6A的特征峰的吸收强度基本不变,据包合前后主客体特征峰波数与吸收强度的变化,可推断出成功制备了GK/PP6A 包合物。

图2 PP6A、芫花素、GK/PP6A 包合物的FT-IR 吸收光谱图Fig.2 FT-IR absorption spectra of PP6A, genkwanin, and GK/PP6A inclusion complex
2.3.2 XRD 分析 使用X 粉末射线衍射仪对样品进行XRD 测试,测试条件为Cu Kα(λ=0.154 06 nm),2θ扫描区间从10°~90°,管电压40 kV,管电流40 mA,扫描速率10°/min。PP6A、芫花素和GK/PP6A 包合物的XRD 如图3 所示,PP6A 在2θ值为13.13°和15.16°处有2 个弱的衍射峰,在19.24°~21.31°有1 个弱的宽衍射峰,呈现出明显的无定形结构。芫花素呈现出良好的晶型结构,在2θ值为10.79°、14.16°和18.66°处有较强且尖锐的衍射峰,且在12.21°、16.25°、21.33°、24.55°、26.90°、28.75°和44.19°处也均有明显尖锐的衍射峰,当芫花素与PP6A 形成包合物后,显示出了不同于主体或客体的衍射模式,包合后PP6A 与芫花素的特征衍射峰基本消失,此外,GK/PP6A 包合物在21.95°和31.23°处出现了较强且尖锐的衍射峰。实验结果表明,主客体包合前后其晶型结构、衍射峰位置和强度均有变化,说明包合物成功制备。
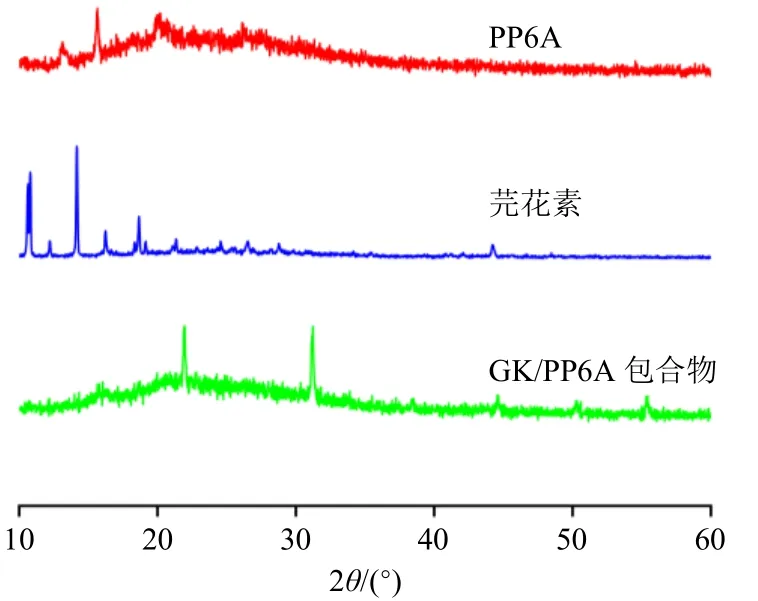
图3 PP6A、芫花素、GK/PP6A 包合物的XRD 图Fig.3 XRD patterns of absorption spectra of PP6A,genkwanin and GK/PP6A inclusion complex
2.3.3 SEM 分析 采用NOVA NANOSEM-450 型扫描电子显微镜对样品的表面形态进行扫描,将样品均匀的分散在金属样品台上,并在真空中喷金处理。在30 kV 的激发电压下拍摄得到SEM 图像。SEM 是物质表面结构定性分析最常用的方法之一,PP6A、芫花素和GK/PP6A 的SEM 图如图4 所示,由SEM 测试结果可知,PP6A 的表面形态平整光滑且均匀分布着细碎的小斑点,形状为规则的片状;芫花素的表面形态呈现为细长的针状,形似一簇堆叠的松针;GK/PP6A 的表面形态呈现出表面略粗糙且带有条状纹理的块状结构,包合前表面细碎的小斑点基本消失,呈现出与前两者不同的表面形态。包合前后表面形貌的变化,初步揭示了GK/PP6A 包合物的形成。

图4 PP6A、芫花素、GK/PP6A 包合物的SEM 图像 (SEM 图经Photoshop 染色)Fig.4 SEM images of PP6A, genkwanin and GK/PP6A inclusion complex (SEM image stained by Photoshop)
2.3.4 DSC 分析 采用Netzsch STA 449F3 型差示扫描量热仪,设置升温速率为10 ℃/min,温度范围35~150 ℃,氩气体积流量为240 mL/min,对PP6A、芫花素和GK/PP6A 包合物进行差示扫描量热测定,得到DSC 曲线。如图5 所示,PP6A 在76.76 ℃和212.09 ℃分别有1 个吸热峰,在560.89 ℃有1 个放热峰。芫花素在285.10 ℃和408.29 ℃分别有1个吸热峰,在600.92 ℃有1 个放热峰。当芫花素与PP6A 形成包合物以后,芫花素与PP6A 的特征峰均有减弱或消失,且在464.27 ℃和484.67 ℃出现了新的放热峰,515.21 ℃出现了新的吸热峰。DSC 曲线分析结果表明,芫花素与PP6A 已形成包合物。

图5 PP6A、芫花素、GK/PP6A 包合物的DSC 图Fig.5 DSC curves of PP6A, genkwanin and GK/PP6A inclusion complex
2.4 NMR 分析
采用BrukerAvance DRX 400 MHz 核磁共振波谱仪测试了芫花素、PP6A 和GK/PP6A 的1H-NMR、13C-NMR 和2D NMR 谱图。其中,芫花素采用DMSO-d6作为溶剂,PP6A 和GK/PP6A 均以D2O作为溶剂,氘代溶剂均以四甲基硅烷(TMS)作为内标。
2.4.11H-NMR 分析 核磁共振波谱是研究主客包合物的有效方法[28]。芫花素与PP6A 进行主客体络合时,化学位移会随着主客体的相互屏蔽作用发生变化,因此可通过主体在包合前后质子的化学位移变化来推断客体分子是否顺利进入主体空腔中。PP6A和GK/PP6A的1H-NMR如图6所示,GK/PP6A包合物的1H-NMR 同时包含了主客体分子的特征质子峰,且包合前后PP6A 的特征质子峰H1、H2、H3、H4的质子信号分别均向低场移动了δ0.05、0.05、0.04、0.11,结果如表4 所示,核磁共振波谱测试结果表明成功制备了包合物。

表4 PP6A 包合前后的化学位移值 (ΔδH) 对照Table 4 Comparison of chemical shift (ΔδH) values before and after PP6A inclusion
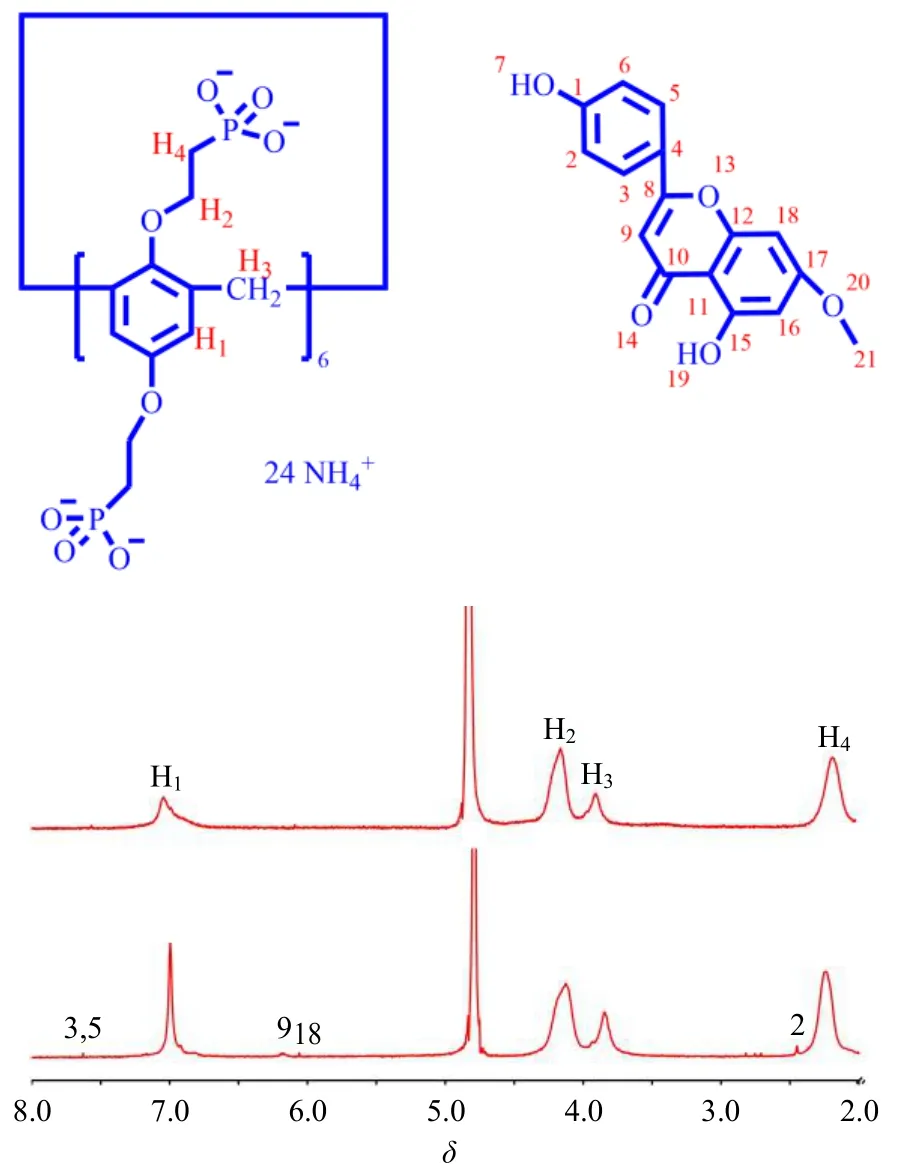
图6 PP6A 与GK/PP6A 的1H-NMR 谱图Fig.6 1H-NMR spectra of PP6A and GK/PP6A
2.4.2 2D-NMR 分析 主客体络合时空间邻近的2个质子产生的核奥弗豪泽效应(nuclear Overhauser effect,NOE)能有效证明主客体之间是否发生络合,采用二维核磁共振波谱对样品进行分析,结果如图7 所示,测试结果表明PP6A 的H-1 与芫花素的H-2、H-9 存在NOE 相关,PP6A 的H-2 与芫花素的H2 存在NOE 相关,说明芫花素成功进入PP6A 的空腔内形成包合物。
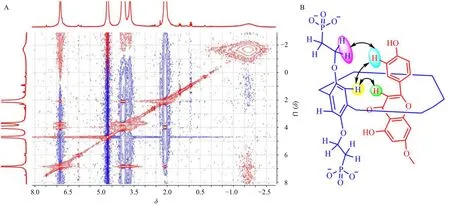
图7 GK/PP6A 包合物的2D NOESY 谱 (A) 及其可能的包合模式和关键的NOE 相关 (B)Fig.7 Two-dimensional nuclear Overhauser effect spectroscopy (2D-NOESY) spectrum (A) and proposed inclusion modes associated with key nuclear Overhauser effects (NOEs) (B) of GK/PP6A inclusion complex
2.5 光谱测定
2.5.1 Job 曲线测定 采用等物质的量连续变化法测定GK/PP6A 的包合计量比[29],维持体系总浓度为10.0 μmol/L,保持芫花素与PP6A 的物质的量比为0∶10、1∶9、2∶8、3∶7、4∶6、5∶5、6∶4、7∶3、8∶2、9∶1、10∶0,并配制系列浓度梯度的混合溶液测定紫外吸收光谱,以芫花素的摩尔分数为横坐标,A值之差与芫花素的摩尔分数乘积为纵坐标,绘制得Job 曲线。如图8-a 所示,当芫花素的摩尔分数为0.5 时,溶液的A值之差与芫花素的摩尔分数乘积达到最大。由此可以得出GK/PP6A 的包合物质的量比为1∶1。

图8 PP6A 与芫花素的Job 曲线 (a) 和PP6A 与芫花素的荧光滴定曲线 (b)Fig.8 Job curve between PP6A and genkwanin (a) and ultraviolet titration curves of PP6A and genkwanin (b)
2.5.2 紫外-可见光谱滴定实验分析 通过紫外-可见吸收光谱(UV-Vis)法探究了芫花素与PP6A 间的主客体络合行为,如图8-b 所示,保持芫花素的浓度为10.0 μmol/L,按照等浓度梯度依次加入0~20.0 μmol/L 的PP6A 溶液,测定紫外吸收光谱。由测试结果可知,随着PP6A 溶液的加入混合溶液的A值逐渐增强,说明芫花素与PP6A 之间存在着较强的主客相互作用。根据滴定过程中A值的变化,采用Benesi Hildebrand 曲线法[30]求解得芫花素与PP6A 的络合常数为49 488 L/mol。
2.6 芫花素与PP6A 的主客体包合模拟计算分析
采用Gaussian 03 程序[31]完成量化计算,PP6A的初始结构由Gaussian View 对全甲氧基柱[6]芳烃(取自剑桥晶体数据库)进一步修饰得到,并用PM6-D3H4 方法对PP6A 的初始结构进行优化至结构达到几何平衡,同时采用Gaussian View 构建芫花素的初始结构至结构达到几何平衡。
采用MOPAC2016 软件包进行半经验计算,选用坐标放置法构建芫花素与PP6A 的包合过程:以芫花素分子上13 号位置的氧原子作为定位点m,以PP6A 上连接的亚甲基上的碳所构成圆的几何中心作为坐标原点O,并将m 与O 之间的距离定义为Dm-o,使芫花素分子从距离坐标原点的10 nm 移动到−10 nm,每步移动1 nm,且均用PM6-D3H4 方法对包合物结构进行全优化。
采用AutoDock 4.2 程序[32]对芫花素与PP6A 的包合模式进行深入探究并完成分子对接。将芫花素和PP6A 分别作为配体分子和刚性受体分子进行分子对接模拟,设置最大评估数为2.5×106,执行100次构像搜索,将芫花素对接到PP6A 的空腔中,并将构像搜索过程中均方根偏差(root mean square deviation,RMSD)小于0.2 nm 的构像归为一簇,进行聚类分析。芫花素以每步1 nm 的步长靠近并深入PP6A 的空腔中,整个过程的结合能变化曲线如图9 所示,由计算结果可知,随着Dm-o减小,当芫花素移动到3 nm 时,结合能为−105.184 4 kJ/mol,此时结合能力达到最强,此位置对应的GK/PP6A 络合体系的结合模式如图10-A 所示。

图9 芫花素与PP6A 的结合能变化曲线图Fig.9 Diagram of binding energy variation between genkwanin and PP6A

图10 经PM6-D3H4 优化后的最优GK/PP6A 包合物模式(A) 和GK/PP6A 包合物分子对接最优构像 (B)Fig.10 Complexation patterns of optimal GK/PP6A inclusion complex after PM6-D3H4 optimization (A) and optimal molecular docking conformation of GK/PP6A inclusion complex (B)
当芫花素移动到3 nm 时,客体芫花素置于PP6A 的空腔中,二者空间匹配度良好紧密结合,与二维核磁共振氢谱推断的主客体可能的包合模式相互吻合。随着芫花素继续移动至4~−6 nm 间时,由于PP6A 支链和芫花素分子的扭转,芫花素分子未能较好的置于PP6A 的空腔中,自−7 nm 后,随着芫花素继续移动且即将脱离PP6A 的空腔时,因为空间斥力的减弱,结合能力呈现出逐渐减弱的趋势。值得注意的是,采用AutoDock 4.2 程序对芫花素与PP6A 的包合模式进行深入探究并完成分子对接后,得到GK/PP6A 包合物分子的对接最优构像如图10-B 所示,并发现芫花素分子7 号位上的羟基暴露于PP6A 的空腔外,并与PP6A 中苯环对位的醚氧原子形成了1 条氢键,长度为0.186 6 nm,主客体间紧密结合并形成氢键,进一步增强了主客体间的结合能力。分子对接完成后,将RMSD 小于0.2 nm 的构像归为一簇,进行聚类分析,芫花素和PP6A 的构像聚类分析如表5 所示,分析结果表明对接结果收敛度较高,只有五簇形成。

表5 芫花素与PP6A 分子对接后的成簇分析(RSDGK/PP6A<0.2 nm)Table 5 Clustering analysis diagram of genkwanin and PP6A molecules after docking (RSDGK/PP6A < 0.2 nm)
2.7 包合物的性能研究
2.7.1 包合物的溶解度测定实验 通过饱和溶液的方法测定芫花素和GK/PP6A 包合物在水中的溶解度,分别制备芫花素和GK/PP6A 包合物的饱和水溶液,并通过芫花素的紫外吸收标准曲线分别计算出芫花素包合前后的溶解度。实验结果表明,芫花素包合前的溶解度为3.5 μg/mL,包合后的溶解度为540.5 μg/mL,约提高了155 倍,由此可知PP6A 能有效的改善芫花素的溶解度。
2.7.2 包合物的稳定性实验 胃和肠道是人体吸收口服药物的主要场所,因此模拟生物体环境对芫花素和GK/PP6A 的稳定性进行探究[33]。设置2 组实验,每组各取2 个25 mL 量瓶,分别向瓶中依次加入0.01 mmol/L 的芫花素溶液和4 mmol/L 的PP6A溶液,用pH 1.5(模拟人体胃液)和pH 7.6(模拟人体肠液)的缓冲溶液与甲醇的混合液(4∶1)分别稀释至刻度,37 ℃恒温孵育1 h,于最大吸收波长处测定其A值,然后避光保存,每隔12 h 测定其A值,连续测定72 h,每组实验平行测定3 次。芫花素和GK/PP6A 的稳定性如图11 所示,在模拟人体的胃液环境中,芫花素在0~36 h 的相对A值呈现出明显的下降趋势,至36 h 下降了85.45%,在模拟人体的肠液环境中,芫花素在0~24 h 的相对吸光度(A/A0,A为每12 h 内所测吸光度,A0为初始吸光度)呈现出明显的下降趋势,至24 h 下降了96.51%,而GK/PP6A 包合物的相对A值在模拟的胃液与肠液的环境中均表现为下降趋势平缓。实验结果表明芫花素在形成包合物后稳定性显著提高。

图11 芫花素和GK/PP6A 包合物在pH 1.5 和pH 7.6 的条件下的A/A0 随时间变化图Fig.11 Plot of relative absorbance A/A0 of genkwanin and GK/PP6A inclusion complex with time at pH 1.5 and pH 7.6
3 讨论
选取PP6A 作为药物载体,芫花素为客体分子,构筑了一种新型的GK/PP6A 超分子包合物。以包合物的包合率为指标设计正交试验,得GK/PP6A 包合物的最优制备工艺为PP6A 与芫花素的物质的量比为1∶1,包合温度为50 ℃,包合时间8 h,甲醇与超纯水体积比为1∶8,包合率为89.93%。并探究了4 个因素在不同水平间的显著性差异,包合时间在不同水平间存在显著性差异,是包合物的制备工艺的重要影响因素。
采用核磁共振波谱、FT-IR、XRD、SEM 和DSC等分析测试方法对包合物进行了表征,主客体在包合前后其理化性质发生了明显变化。通过半经验分子轨道法和分子对接揭示了芫花素与PP6A 最稳定的包合模式,疏水作用和氢键作用为包合物形成的主要驱动力。进一步通过紫外可见吸收光谱探究了芫花素和PP6A 的物质的量比及络合常数,试验结果表明,芫花素和PP6A 以物质的量比1∶1 形成超分子包合物,其络合常数为49 488 L/mol,芫花素与PP6A 之间存在着较强的主客相互作用。包合物的性能研究实验结果表明,芫花素与PP6A 形成超分子包合物后,水溶解性和稳定性显著提高,其中水溶解性由3.5 μg/mL 提升至540.5 μg/mL,约提高了155 倍,PP6A 是芫花素的优良药物载体。
综上所述,该研究为天然药物分子芫花素在临床上药物制剂的研究和制备奠定了一定的基础。
利益冲突所有作者均声明不存在利益冲突
猜你喜欢
西藏艺术研究(2021年3期)2021-06-02
马克思主义哲学研究(2020年2期)2020-07-21
中成药(2018年8期)2018-08-29
中成药(2018年5期)2018-06-06
中成药(2017年12期)2018-01-19
首都食品与医药(2017年15期)2017-11-03
中成药(2017年5期)2017-06-13
大观(2017年2期)2017-04-07
科技资讯(2015年19期)2015-10-09
中国现代中药(2014年12期)2014-09-26
