
无铬鞣废革屑胶原提取物与PBAT 共混成膜性能
2024-01-08 07:47侯振强张辉强西怀
皮革科学与工程 2024年1期
侯振强,张辉*,强西怀,2
(1.陕西科技大学轻工科学与工程学院,陕西 西安 710021;2.轻化工程国家级实验教学示范中心,陕西 西安 710021)
为了贯彻“创新、绿色、协调、开放、共享”的新发展理念,对于在皮革生产和成品革加工中产生的固体废弃物[1-2],价值化利用和避免二次污染已成为制革相关领域的研究热点[3]。
当前制革固体废弃物提取胶原的研究包括含铬废革屑脱铬提取[4-9],以及无铬鞣革中胶原、多肽的提取及高值利用[10-13]。除了传统无铬鞣产生的废革屑,一些新型无铬鞣产品在国内市场陆续出现且在皮革生产中所占比例逐渐增大,关于新型无铬鞣废革屑的胶原提取特性及相关利用还有待研究。TWS 是由四川亭江新材料有限公司开发生产的一种绿色环保的新型有机鞣剂,鞣剂分子通过烯胺键和酰胺键与胶原分子形成多点交联[14]。荷兰Smit&Zoon 公司生产的沸石鞣剂,其铝组分在酸性条件下可与胶原的天冬氨酸、谷氨酸侧链羧基配位结合,而硅组分通过氢键和胶原羟基发生反应,在纤维之间生成三维网络结构[15]。
目前胶原水解物提取的常用方法中,碱法提取得到的相对分子质量较小,酶法提取对条件控制的要求较高,稀酸法提取得到的胶原水解物纯度高[16]。明胶作为胶原蛋白部分变性的产物,其制备的明胶膜通常具有良好的力学性能、优异的氧气阻隔性能以及良好的生物相容性。目前从废弃物中提取明胶制备聚合物薄膜的研究主要分为两类,一类是从家禽、鱼皮和未鞣制牛皮废料或副产品中提取明胶,并用于生产包装复合薄膜或食用级涂层[17-19],另一类则是从鞣后皮革废料中提取,并应用于农业地膜[20-23]。聚己二酸对苯二甲酸丁二醇酯(PBAT)被认为是制造可生物降解包装材料、堆肥袋和农用地膜最有前景的材料之一[24]。但PBAT 膜存在抗拉强度低、氧气阻隔性能差、成本高以及降解速率慢等缺点。因此通过将PBAT 与其他材料复合,改善PBAT 性能且降低成本已成为当前研究热点问题[25]。
本文以稀酸法提取TWS 鞣废革屑胶原水解物(C-TWS)和沸石鞣废革屑胶原水解物(C-Zeolite),对最适工艺得到的产物进行纯化,旨在得到相对分子质量较高且矿物盐含量低的胶原水解物。研究不同鞣剂结合机理对提取工艺及胶原水解物性质的影响。对纯化胶原水解物进行相关性能表征,探究其作为“无铬”蛋白质生物基材的基本特性,为无铬鞣胶原的工业化提取及进一步拓展应用做基础性工作。选取C-TWS 复合PBAT,并探究PBAT 与胶原水解物的相容性,为生物可降解包装膜和地膜的研究提供参考。
1 实验部分
1.1 实验材料和仪器
1.1.1 主要材料
沸石鞣废革屑和TWS 鞣废革屑取自兴业皮革股份有限公司;硫酸、盐酸、硝酸、硼酸、十二烷基硫酸钠(CP)、四硼酸钠,国药集团化学试剂有限公司;硫酸铜、无水硫酸钾,天津欧博凯化工有限公司;L-亮氨酸,上海浩鸿生物医药科技有限公司;邻苯二甲醛、二硫代苏糖醇,上海麦克林生化科技有限公司;PBAT,新疆屯河801T;2,2,2-三氟乙醇,山东科源生化有限公司。
1.1.2 主要仪器
ZHWY-110 智能数显多功能恒温水浴箱,上海智城分析仪器制造有限公司;1836 乌氏黏度计,合肥申谊玻璃制品有限公司;凯氏定氮仪,实验室自制;FC-10A-E 真空冷冻干燥机,河北国辉实验仪器有限公司;RE52CS 旋转蒸发器,上海市亚荣生化仪器厂;Biochrom30+氨基酸自动分析仪,英国百康有限公司;STA449F3-1053-M 同步热重(TG)-差示扫描量热(DSC)分析仪,德国耐驰仪器制造有限公司;Zetasizer Nano ZSP 纳米粒度表面电位分析仪,英国马尔文仪器有限公司;Vertex70 红外光谱仪、D8 Advance X 射线衍射仪,德国布鲁克仪器公司;W3/060 水蒸气透过率测试仪,济南兰光机电技术有限公司;Cary5000 紫外- 可见- 近红外分光光度计,美国安捷伦科技有限公司。
1.2 实验方法
1.2.1 提取前预处理
称取一定量精细粉碎的废革屑皮粉,设置液固比20 并加入废革屑干质量1%的洗洁精,常温下浸泡12 h,期间搅拌3~4 次,清水洗涤至清洗液中无大量泡沫,纯水洗涤后烘箱干燥备用。
1.2.2 不同鞣制革屑提取条件对比探究
分别称取两种革屑,浓硫酸用量分别为2%、4%、6%、8%、10%、12%(以干革屑计),浸泡12 h,每隔3 h 充分搅拌一次,酸浸结束后直接进行提取。在确定最适酸浸条件的情况下,通过单因素实验探究提取温度(50、60、70、80、90 ℃)、液固比(6、10、14、18、22)、提取时间(2、4、6、8、10 h)对胶原水解物的特性粘数、提取率、灰分等指标的影响,提取液经过滤、离心、中和、浓缩、冷冻干燥后得到粉状胶原水解物。
1.2.3 纯化胶原水解物复合PBAT 制备薄膜
使用截留相对分子质量3 500 Da 的透析袋对胶原水解物进行纯化,以2,2,2-三氟乙醇(TFEA)和少量水为共溶剂,甘油为塑化剂,分别称取一定质量C-TWS、PBAT 配置成5%的溶液,50 ℃下搅拌30 min使各组分充分溶解,然后按照不同组分比例PBAT/C-TWS(90/10、70/30、50/50、30/70、10/90)混合1 h,共混结束后置于40 ℃水浴中超声消泡30 min,浇铸于聚四氟乙烯成膜板,常温条件下干燥成膜。
1.3 分析检测方法
1.3.1 胶原水解物基本特性分析
胶原水解物灰分的测定,按照文献[26]所述方法进行,提取率按照公式(1)进行计算。
式中:W为提取率,%;m1为胶原水解物干质量,g;m2为革屑干质量,g。
室温条件下利用乌氏黏度计测定并推算出革屑提取液的特性粘数[η],相对黏度和增比黏度的计算方式见公式(2)、(3),再由lnηr/C-C及ηsp/C-C作图外推[27]得到[η]。根据Mark-Houwink 方程,将[η]带入标准公式[η]=1.196×10-3×M0.7257,即可得到不同提取条件下胶原水解物的粘均相对分子质量M[28]。
式、中:ηr为相对黏度;ηsp为增比黏度;t为胶原水解物溶液流出时间,s;t0为溶剂流出时间,s。
总氮含量以及粗蛋白含量计算方法如式(4)、(5)所示;选用便捷且耗时短的邻苯二甲醛法测定蛋白质水解度(DH)[29],DH 指的是蛋白质分子断裂的肽键占蛋白质分子中总肽键的比例,即每断裂一个肽键就可同时生成一个游离的氨基和羧基[30],DH的计算公式如式(6)所示。
式中:ω为含氮量,%;N为标准盐酸溶液的浓度,mol/L;V1为滴定空白时消耗盐酸标准溶液的体积,mL;V2为滴定试样时消耗盐酸标准溶液的体积,mL;m为蛋白粉的质量,g;V' 为试样消化液蒸馏用体积,mL;V为试样消化液总体积,mL;P1为氮含量,%;P2为粗蛋白含量,%;C1为提取液游离氨基浓度,mol/L;C2为水解液氮含量,mol/L。
1.3.2 纯化胶原水解物结构表征
设置波数范围400~4 000 cm-1、分辨率4 cm-1,对胶原水解物进行红外光谱分析[31];在氨基酸自动分析仪上采集20 μL 样品进行氨基酸谱分析[32];使用Zeta电位仪分析胶原水解物的等电点;以5 ℃/min 的升温速率在40~70 ℃温度范围内进行DSC 分析;使用扫描电子显微镜能谱仪对材料进行元素成分分析。
1.3.3 PBAT/C-TWS 复合膜性能表征
通过傅里叶变换红外光谱仪分析PBAT/C-TWS 共混膜的分子结构及其化学键;通过TG 分析仪在30~600 ℃测试共混物薄膜的热稳定性;在38 ℃和相对湿度为90%的条件下用水蒸气透过率测试仪测定薄膜的透湿性;通过紫外-可见-近红外分光光度计测定共混物薄膜的光学性质;使用X-射线衍射仪对薄膜进行结构分析,扫描范围为2θ=5~50°。
2 结果与讨论
2.1 革屑理化性质分析
将预处理后TWS 鞣、沸石鞣废革屑烘干至一定程度,其理化性质如图1(a)所示。两种革屑的粗蛋白含量一致,其中TWS 鞣废革屑的水分含量比鞣废革屑大3.26%,但灰分含量要小3.18%,原因可能是由于沸石鞣剂在鞣制过程中引入的灰分较大。

图1 废革屑理化性质(a)和ηsp/C,lnηr/C 与C 的关系曲线(b)图Fig.1 Physicochemical properties of waste leather scraps(a)and the curves of ηsp/C,lnηr/C and C(b)
2.2 提取液特性粘数分析
以单因素实验中酸用量8%、温度70 ℃、液比1∶10、提取时间6 h 得到的C-TWS 纯化样品为例,乌氏黏度计溶剂流出时间t0=128.85 s,待测液浓度与流出时间及相关粘数如表1 所示,以溶液的浓度C为横坐标,以ηsp/C及lnηr/C为纵坐标做出ηsp/C-C及lnηr/C-C的关系图,通过外推法得到的特性粘数[η]为6.78,如图1(b)所示。
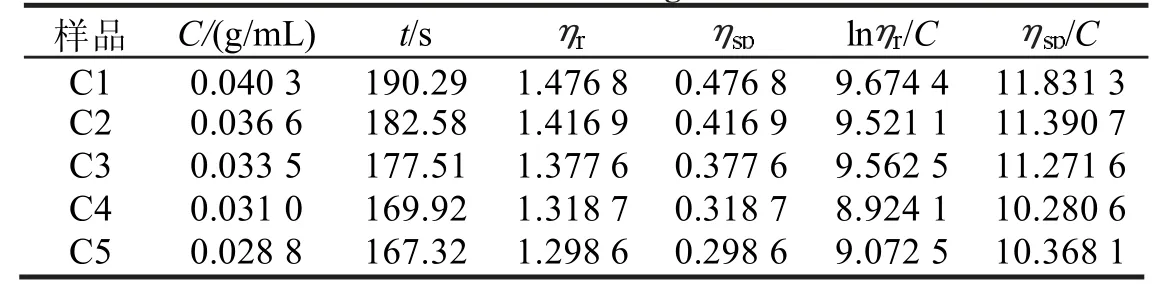
表1 不同C-TWS 浓度梯度下蛋白质流出时间及相应数值Tab.1 Protein’s efflux time and the corresponding values at different CTWS concentration gradients
2.3 提取条件分析及工艺优化
不同酸用量提取得到胶原水解物的性质如图2(a)所示,当硫酸用量8%时C-TWS 提取率达到最大值96.84%,C-Zeolite 提取率在硫酸用量8%时可达76.74%。随着酸用量的增加,革屑不断吸水膨胀,胶原结构中的一部分共价键断裂,从而促使胶原水解物的释放,表明酸浸浓度的增大更利于后续升温提取胶原水解物的不断溶出。两种胶原水解物的水解度随着酸用量的增加而持续增大,提取液特性粘数则呈现先增大到峰值而随后减小的趋势。酸用量较少时,反应主要以革屑内部的大分子胶原水解物溶出为主。当酸用量过度时胶原水解度迅速增大,而对应提取液特性粘数不断减小,较大的酸浓度对溶出的胶原水解物起到了降解作用。通过综合考虑不同酸用量得到的胶原水解物各项指标,C-TWS 提取的最适硫酸用量为8%,C-Zeolite 提取的最适硫酸用量为10%。

图2 提取条件对胶原水解物提取率、灰分、水解度、特性粘数的影响(a.酸用量;b.水解时间;c.水解温度;d.液固比)Fig.2 Effect of extraction condition on the extraction rate, ash content, degree of hydrolysis, and characteristic viscosity of the resulting collagen hydrolysates(a.acid dosage;b.hydrolysis time;c.hydrolysis temperature;d.liquid-solid ratio)
分别选取最适酸用量,固定反应温度70 ℃、液固比10,不同时间提取得到胶原水解物的性质如图2(b)所示,对于两种革屑而言,反应时间延长有助于胶原水解物提取率的提升,使得酸对革屑肽键的水解反应更彻底。C-TWS 和C-Zeolite 在反应6 h 和8 h 时提取率分别达到最大值,而后随时间的增加,其提取率皆呈现稳定趋势。表明当反应达到一定程度时,时间过度延长也无法对胶原水解物的提取率产生影响。随着反应时间的延长,大分子胶原水解物被酸逐步降解,到达一定时间后提取液中出现大量小分子多肽,故水解度增加趋势减缓。同时提取液特性粘数也随着时间的延长不断减小,时间过长则趋于稳定趋势。
固定各自最适酸用量及反应时间,设置液固比10,不同反应温度提取得到胶原水解物的性质如图2(c)所示。两种胶原水解物的提取率都随着反应温度的升高而不断增加,在温度升高至70 ℃时,C-TWS 提取率达到最大值96.31%,80 ℃时C-Zeolite 提取率达到最大值79.66%。升温有助于提取过程中吸热反应的进行,温度的升高在一定程度上加快了分子运动,加强了分子之间的碰撞,从而增加了胶原水解物的提取率。两种胶原水解物的水解度呈逐渐上升趋势,而特性粘数逐渐减小。证明较低温度不利于反应的进行,温度的升高促进肽键断裂,而升高到一定程度后,分子运动达到极限,提取率和水解度趋于平稳。
TWS 鞣革提取条件设置为酸用量8%、反应时间6 h、反应温度70 ℃,沸石鞣革提取条件设置为酸用量10%、反应时间8 h、反应温度80 ℃,探究不同液固比提取得到胶原水解物的性质如图2 (d)所示。两种胶原水解物提取率均随着液固比的增加呈先升后降,之后趋于平稳的趋势。当液固比较小时,体系中革屑所占比重较大,革屑与酸液不能够充分接触,故增加液固比即是增加屑水解的反应面积,有利于提取率的提高。但当液固比过大时,会导致酸浓度降低,从而降低了反应效率。由图中提取率及灰分趋势可知,提取C-TWS 及C-Zeolite 的最适液固比都是10。随着液固比的增加,两者胶原水解度均呈先增大后减小的趋势,液固比较小的情况下,革屑在提取过程中得不到充分浸泡,增大液固比可以引起明显的特性粘数变化,当革屑与提取酸液的比例达到10,多余的水会降低胶原水解物的水解度,使特性粘数基本不再改变。
2.4 胶原水解物的纯化分析
皮革生产过程中使用大量无机盐,以及鞣剂引入的不同离子组分使得废革屑灰分较大,且利用硫酸提取胶原水解物的过程中又会生成一部分硫酸盐。纯化工艺旨在减小灰分和若干金属离子对胶原产品带来的负面影响,保留较大相对分子质量的胶原水解物,并提高其纯度。C-TWS 及C-Zeolite 纯化前后灰分变化如图3(b)所示,灰分含量大幅度降低且达到工业明胶(QBT 1995-2005)灰分指标,主要元素含量变化结果见图3(c),纯化前后水解物中的矿物盐含量明显降低,尤其以S、Cl、Na 元素含量变化最为显著。

图3 胶原水解物纯化效果及性能表征(a.革屑及纯化胶原水解物;b.纯化前后灰分变化;c.纯化前后主要元素含量变化;d.Zeta 电位测试;e.红外图谱;f.DSC 曲线;g.氨基酸组成及含量;h.圆二色谱图)Fig.3 Purification effect and performance characterization of collagen hydrolysate(a. Leather chips and purified collagen hydrolysate; b. ash content change before and after purification; c. content changes of major elements before and after purification;d.Zeta potential test results;e.infrared spectra;f.DSC curves;g.amino acid composition and content;h.circular dichroism curves)
2.5 纯化胶原水解物性能表征
如图3(d)可知C-TWS 等电点约为5.18,而C-Zeolite 等电点约为7.34。未鞣制酸皮胶原纤维的等电点基本在5.2~5.4[34],C-TWS 等电点偏酸性,是由于TWS 鞣剂自身的阴电性较强,而沸石鞣剂与胶原侧链羧基基团的配位结合导致坯革纤维上的负电性基团减少,故C-Zeolite 的等电点要高于C-TWS。
C-TWS 及C-Zeolite 红外图谱如图3(e)所示,两种胶原水解物的酰胺Ⅰ带在1 600~1 700 cm-1区域的强烈吸收指的是蛋白多肽骨架上C=O 的伸缩振动。酰胺Ⅱ带在1 550 cm-1附近有中强吸收,一般指N-H 的平面弯曲和C-N 伸缩振动的组合[35]。在1 220~1 330 cm-1处是红外吸收区域为酰胺Ⅲ带,主要反应酰胺键内的C-N 伸缩振动和N-H 平面弯曲,也和甘氨酸、脯氨酸侧链的CH2基团产生的震动吸收有关,由此可见两种胶原水解物中均存在多肽成分。两种胶原水解物的酰胺A 都出现在3 400~3 440 cm-1波数范围内,表明存在N-H 键的伸缩振动。2 870~3 000 cm-1的吸收峰表明两者皆存在酰胺B,通常指的是CH2的不对称伸缩振动,C-TWS 在2 934 cm-1处的吸收峰表示存在C=N 双键的弯曲震动和共振振动,表明TWS 鞣剂与胶原结合的烯胺键未被全部水解。C-TWS 和C-Zeolite分别在1 450 cm-1和1 452 cm-1处出现的吸收峰,与脯氨酸、羟脯氨酸的吡咯烷环振动有关。区别于C-TWS,C-Zeolite 在1 153 cm-1处有明显的强吸收峰,说明该体系中存在着硅氧键的伸缩振动。
图3 (f)为C-TWS、C-Zeolite 纯化后冻干样品的DSC 曲线,胶原蛋白的热转变是一个非瞬时的吸热过程。随着外界温度不断升高,胶原蛋白的三螺旋结构先被部分破坏进而全部破坏,达到胶原变性的目的。C-TWS、C-Zeolite 的热变性温度分别为66.1 ℃和58.6 ℃。C-TWS 热变性温度过高,可能是由于其胶原结构中依旧存在少量稳定的烯胺键,与红外谱图分析结果相同。
C-TWS、C-Zeolite 氨基酸组成及含量如图3(g)所示,两种胶原水解物均含有17 种氨基酸,其中C-TWS 氨基酸总量占比97.85%,C-Zeolite 氨基酸总量占比94.89%。对比高贵贤[36]从原皮中提取的胶原蛋白,本实验提取得到的两种胶原水解物氨基酸含量均偏小,且种类略少。两种样品中,胱氨酸、酪氨酸、组氨酸的含量都相对较低,甘氨酸、脯氨酸、谷氨酸和天冬氨酸含量较高。
Zhang[37]等对同种原料制备的胶原蛋白、明胶、胶原水解物的圆二色谱进行分析,得出胶原蛋白在221 nm 处有一个正最大峰,在192 nm 处有一个负最小峰,表明其具有典型的三螺旋构象,而明胶与胶原水解物的三螺旋结构均转变为无规卷曲构型。图3(h)为C-TWS、C-Zeolite 的圆二色谱图,其中胶原水解物在221 nm 处没有正峰,显示出无规卷曲构型,表明两种纯化胶原水解物已经不具备三螺旋空间构象。
2.6 PBAT/C-TWS 复合膜性能表征
薄膜的FT-IR 分析如图4 (a) 所示,在PBAT/C-TWS 复合膜的红外谱图中,3 311 cm-1处出现的宽吸收峰归因于胶原水解物的-NH 和-OH伸缩振动,与图3(e)中C-TWS 红外图谱对比,-NH和-OH 的相应波数降低,说明薄膜基质中存在较强的氢键等分子间相互作用力。1 712 cm-1的吸收峰归因于PBAT 中C=O 的伸缩振动,1 255 cm-1和1 107 cm-1处的峰带归因于C-O 键的伸缩振动,723 cm-1处的峰与PBAT 中多个相邻甲基的振动有关[24]。复合膜红外谱图无新化学键产生,证明PBAT与C-TWS 成膜过程为单纯的物理共混。
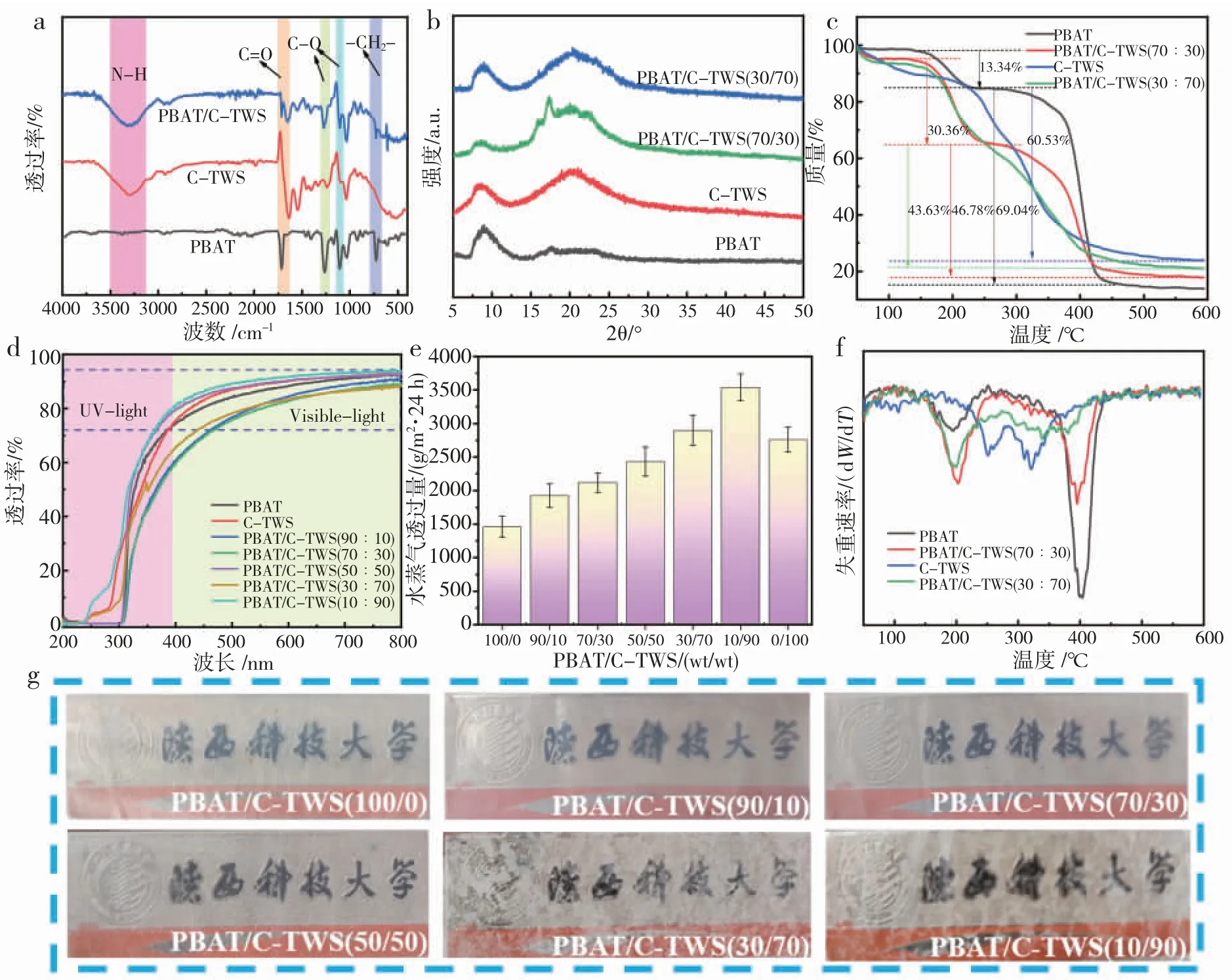
图4 PBAT/C-TWS 复合膜性能表征(a.红外图谱;b.XRD 图谱;c.TG 曲线;d.紫外-可见透光率曲线;e.水蒸气透过量;f.DTG 曲线;g.透光性图像演示)Fig.4 Performance characterization of PBAT/C-TWS composite film(a.IR spectra;b.XRD spectra;c.TG curves;d.UV-visible transmittance curves;e.water vapor transmission coefficients;f.DTG curves;g.Light transmittance image demonstration)
不同PBAT 和C-TWS 比例制备的复合膜X-射线衍射图谱如图4(b)所示,当共混体系中PBAT 含量大于C-TWS 含量时,复合膜在17.4°的特征衍射峰比较明显,而胶原水解物膜的特征衍射峰在20.5°附近,随着共混体系中C-TWS 组分的增加,PBAT 特征峰强度减弱,可能是分子间的作用力干扰了PBAT 晶格的有序性。与单体的特征峰相比,共混体系中未出现新的峰,且特征峰的位置几乎没有发生偏移。
不同PBAT 和C-TWS 比例复合膜的TG 和DTG 曲线如图4(c、f)所示,所有膜显示出两个降解过程,第一个降解过程为甘油,其失重温度范围为189~240 ℃。当C-TWS 组分含量为0 时,PBAT 的最大热降解温度为405.4 ℃;C-TWS 组分含量为30%时,PBAT 的最大热降解温度降低至386.5 ℃。随着C-TWS 含量的增加,复合膜在热降解初期的失重率增加,说明C-TWS 的加入降低了复合膜的热稳定性。
薄膜的光学性能在包装和农业地膜领域的应用具有重要影响,其透光率曲线如图4(d)所示,所有薄膜均具有较高的可见光透明度。随着C-TWS 含量的增加,复合膜的透光率呈现先减小后增大的趋势,原因可能是C-TWS 的含量达到一定程度时,其在体系中由分散相转变为连续相,与PBAT 的相界面减少,从而提高了复合膜的透光率。同时由图4(g)可知,当C-TWS 组分含量达到70%和90%时,复合膜显示出明显的不均匀表象,共混体系出现了相分离。
水蒸气透过量(WVT)同样是薄膜选择的重要特性。如图4(e)所示,纯PBAT 薄膜的WVT 值为1 458.3 g/(m2·24 h);随着C-TWS 含量的增加,复合膜的WVT 分别提高了43.05%、78.17%、94.36%、139.57%和182.5%。而C- TWS 薄膜的WVT 值为2 761.6 g/(m2·24 h),小于C-TWS 组分含量70%和90%的水蒸气透过量,可能是共混体系中存在两相界面,说明当成膜体系中C-TWS 组分含量过大时,PBAT 与C-TWS 之间的相分离和较差的相容性更有利于水分子在复合膜中的渗透。
3 结论
C-TWS 的最适提取工艺为:硫酸用量8%,温度70 ℃,液固比10,时间6 h。C-Zeolite 最适的水解工艺为:硫酸用量10%,温度80 ℃,液固比10,时间8 h。同种提取条件下,C-TWS 的提取率及相对分子质量更大,提取更容易。C-Zeolite 所含灰分较大,且提取液分离困难。两者纯化后的胶原水解物矿物盐含量低,氨基酸含量达到17 种,热变性温度分别为66.1 ℃和58.6 ℃。可作为绿色蛋白基材,满足无铬鞣废革屑蛋白材料的资源化二次利用。
从红外谱图和XRD 谱图分析可以看出,PBAT与C-TWS 共混成膜过程仅是简单的物理共混。随着胶原水解物在共混体系中的含量不断增大,PBAT/C-TWS 复合膜水蒸气透过量增大,热稳定性不断降低。所有比例复合膜均具有较高的可见光透明度,但C-TWS 组分含量达到70%和90%时,共混体系出现了明显的相分离,证明复合膜组分比例中胶原水解物比例较低时与PBAT 成膜显示出较好的相容性。
猜你喜欢
Journal of Forestry Research(2018年2期)2018-03-19
科学与财富(2017年28期)2017-10-14
西南国防医药(2016年6期)2016-12-01
东华理工大学学报(自然科学版)(2016年1期)2016-05-23
中国塑料(2016年1期)2016-05-17
中国卫生标准管理(2015年15期)2016-01-15
中国塑料(2015年3期)2015-11-27
中国医疗美容(2015年4期)2015-04-27
中国塑料(2014年10期)2014-10-17
食品科学(2013年13期)2013-03-11
