
胶质瘤化疗耐药机制的表观遗传学调控研究进展△
2024-01-04 08:57陈曦王雅梅
癌症进展 2023年20期
陈曦,王雅梅
首都医科大学12021级临床医学“5+3”一体化专业一班,2基础医学院生化与分子生物学系,北京 1000690
胶质瘤是发生于神经外胚层的颅内肿瘤,占脑部肿瘤的70%以上,其隐匿性较强,恶性程度、复发率及病死率均较高[1]。临床上将胶质瘤分为4 个级别,Ⅰ、Ⅱ级胶质瘤称为低级别胶质瘤,Ⅲ、Ⅳ级胶质瘤称为高级别胶质瘤,级别越高,恶性程度就越高。低级别胶质瘤患者不常见,大多为年轻患者,这些患者有较好的预后,并对治疗表现出一定的敏感性[2]。多形性胶质母细胞瘤(glioblastoma multiforme,GBM)是Ⅳ级星形细胞肿瘤,也是病死率最高、最难治愈的肿瘤。高级别胶质瘤的治疗以手术切除为主,放化疗为辅。替莫唑胺(temozolomide,TMZ)是一种口服烷基化药物,也是唯一可以跨越血脑屏障对胶质瘤起作用的药物,可将细胞周期阻断在G2/M 期,导致胶质瘤细胞凋亡[3]。铂类药物如顺铂、卡铂等是一类影响细胞周期的化疗药物,也是临床上治疗胶质瘤的常用化疗药物,其主要靶点是DNA,通过与DNA结合破坏其结构,最终导致胶质瘤细胞周期中断或凋亡[4]。经过多年研究发现,胶质瘤对化疗具有很强的耐药性,这也导致患者治愈率不高,生存期较短,预后较差。越来越多的研究表明,表观遗传学调控因子的改变对胶质瘤的发生发展及耐药具有重要影响。因此,研究胶质瘤的耐药机制,探索减少胶质瘤产生耐药的方法对提高抗肿瘤药物的疗效具有重要的临床意义。本文从胶质瘤耐药的产生机制及表观遗传学调控对胶质瘤耐药的影响等方面进行综述,旨在对胶质瘤的治疗、药物的合理使用和研发以及后续的相关研究提供参考依据。
1 表观遗传学调控对胶质瘤耐药的影响
表观遗传修饰参与多种生命活动的调控过程,常见的修饰类型包括DNA 甲基化、组蛋白修饰、非编码RNA 调控和染色质重塑,表观遗传修饰异常可能诱导胶质瘤发生发展与耐药的产生。
1.1 DNA 甲基化
DNA 甲基化是一种在DNA 甲基转移酶(DNA methyltransferase,DNMT)的催化下进行的化学修饰过程。在这个过程中,以S-腺苷甲硫氨酸(S-adenosyl methionine,SAM)作为甲基供体,在胞嘧啶的嘧啶环上的5'位置添加一个甲基,形成5-甲基胞嘧啶,从而使基因沉默,该过程主要发生在CpG 二核苷酸上,也可见于非CpG 序列,但较为罕见[5]。DNMT 的表达与胶质瘤细胞对TMZ 的敏感性有关。研究发现,DNMT1 在对TMZ 耐药的胶质瘤细胞中表达下调,其表达水平越高,胶质瘤细胞对TMZ 的敏感性越强[6]。DNA 甲基化异常还涉及药物外排转运体的启动子、促凋亡基因和DNA 损伤修复基因。O6-甲基鸟嘌呤-DNA 甲基转移酶(O6-methylguanine-DNA methyltransferase,MGMT)是一种关键的DNA 损伤修复基因,其表达产物负责修复TMZ 造成的DNA 损伤,当MGMT启动子区域发生高度甲基化时,该基因会被沉默,从而增强GBM对TMZ 的临床反应。MGMT 表达上调可使胶质瘤对TMZ 造成DNA 损伤的修复能力增强,从而使胶质瘤产生耐药性。在复发性胶质瘤临床样本中,MGMT启动子容易建立非甲基化状态,MGMT表达出现上调[7]。另一方面,即使MGMT启动子未发生改变,基因组重排列也可导致复发性胶质瘤中MGMT 过表达[8]。Lu 等[9]研究小核仁RNA 宿主基因12(small nucleolar host gene 12,SNHG12)在GBM 耐药中的作用,以及表观遗传学调控对其异常表达的影响,结果发现,在对TMZ 耐药的胶质瘤细胞中,SNHG12 表达显著上调,且启动子处DNA甲基化水平异常低。DNA 甲基化缺失使SNHG12的启动子区域更容易被特异性蛋白1(specificity protein 1,SP1)接近,SP1 可激活SNHG12 并促使其表达。除上述基因外,p17、大肿瘤抑制激酶1(large tumor suppressor kinase 1,LATS1)、大肿瘤抑制激酶2(large tumor suppressor kinase 2,LATS2)等基因启动子区域CpG 岛(基因中富含非甲基化CpG 的区域)的甲基化也与胶质瘤的发生发展和复发耐药密不可分[10-11]。DNA 高甲基化与耐药性有关,因此,去甲基化药物在逆转GBM 中与DNA高甲基化相关的耐药性方面具有潜在的应用价值。但由于缺乏选择性,去甲基化剂可能会产生广泛的全身不良反应,该类药物的临床应用还需要进一步的研究。
1.2 组蛋白修饰
组蛋白是一种蛋白质,可与DNA 形成核小体。核小体是染色质的基本结构单位,是由4 个核心组蛋白(H2A、H2B、H3 和H4)的2 个分子和147个碱基对长度的DNA 形成的组蛋白八聚体。常见的组蛋白表观修饰方式包括乙酰化、甲基化、磷酸化、泛素化和类泛素蛋白修饰等,其中对胶质瘤耐药影响的研究较为深入的是乙酰化[12]。
1.2.1 组蛋白乙酰化修饰对胶质瘤耐药的调控组蛋白乙酰化可以调节染色质结构和基因表达。在这个过程中,组蛋白乙酰转移酶将乙酰基添加到组蛋白上的赖氨酸残基上,从而形成有利于转录激活的开放染色质状态。组蛋白去乙酰化酶(histone deacetylase,HDAC)可以从赖氨酸上去除乙酰基,从而形成有利于转录沉默的致密染色质[13-14]。在GBM 中,一些特殊的HDAC 表达上调,促进了胶质瘤细胞耐药。研究表明,Ⅰ类HDAC的催化活性可促进E3 泛素连接酶RAD18 表达上调,避免细胞发生TMZ 导致的DNA 损伤[15]。Yang 等[16]研究发现,GBM 细胞可以通过HDAC1/2/6 去乙酰化调控SP1的转录活性,使B 细胞特异性莫洛尼鼠白血病病毒插入位点1(B-cell-specific moloney murine leukemia virus insertion site 1,BMI1)和人端粒酶逆转录 酶(human telomerase reverse transcriptase,hTERT)表达上调,并改变多种基因的表达,调节细胞周期G2/M 期和DNA 损伤修复,促进胶质瘤细胞自我更新,从而产生耐药性。组蛋白修饰基因zeste增强子同源物2(enhancer of zeste homolog 2,EZH2)、zeste 12 同源物抑制因子(suppressor of zeste 12 homolog,SUZ12)和AT 丰富结构域1A(ATrich interaction domain 1A,ARID1A)在对TMZ 耐药胶质瘤细胞中的表达水平明显高于对TMZ 不耐药的胶质瘤细胞[17]。与组蛋白修饰相关的酶,如蛋白质精氨酸甲基转移酶(protein arginine methyltransferase,PRMT)和赖氨酸去甲基化酶(lysine demethylase,KDM)均在胶质瘤的发生发展与耐药过程中发挥重要作用,对患者的生存具有负面影响。PRMT 抑制剂目前正在开发中,泛KDM 抑制剂以及选择性赖氨酸去甲基化酶5A(lysine demethylase 5A,KDM5A)抑制剂等小分子抑制剂可有效抑制TMZ耐药胶质瘤细胞继续生长[6]。维持乙酰化平衡状态对于基因转录调节至关重要。未来需要进一步的研究确定HDAC 的最佳组合策略,以确保其在逆转GBM耐药治疗中的有效性和安全性。
1.2.2 胶质瘤的组蛋白修饰调控新机制 随着表观遗传修饰研究的不断深入,很多非乙酰化的组蛋白酰化调控,如组蛋白巴豆酰化修饰、棕榈酰化修饰和琥珀酰化修饰对胶质瘤发生发展影响的研究取得了突破性进展。巴豆酰化修饰主要是在巴豆酰基转移酶的作用下以巴豆酰辅酶A 为底物,将巴豆酰基转移到赖氨酸残基,其修饰位点主要富集在基因的启动子区域和潜在的增强子区域[18]。研究表明,GBM 干细胞通过调节赖氨酸代谢产生大量的巴豆酰辅酶A,从而增加细胞整体巴豆酰化水平。组蛋白H4 的巴豆酰化修饰通过影响H3K27乙酰化和H3K9 三甲基化限制免疫原性转座因子,抑制干扰素信号,减少CD8+T 细胞浸润,最终减弱抗肿瘤免疫反应并促进肿瘤生长[19]。琥珀酰化是指琥珀酰基供体在琥珀酰辅酶A 的介导下将琥珀酰基团共价结合到赖氨酸残基的过程。研究报道,α-酮戊二酸脱氢酶(α-ketoglutarate dehydrogenase,α-KGDH)通过与赖氨酸乙酰转移酶2A(lysine acetyltransferase 2A,KAT2A)结合形成复合物,从而发挥琥珀酰转移酶的功能,进一步使组蛋白H3K79 琥珀酰化,从而促进胶质瘤生长[20]。Zhang 等[21]研究证实了胶质瘤内皮细胞中转胶蛋白2(transgelin 2,TAGLN2)基因K40 位点高度琥珀酰化,在体内外均显著促进胶质瘤血管生成和肿瘤转移。棕榈酰化是一种可逆的蛋白质翻译后修饰,通常指棕榈酰基团与蛋白质的半胱氨酸残基共价连接,形成不稳定的硫酯键。Chen 等[22]研究发现,与正常脑组织相比,胶质瘤组织中棕榈酰化修饰程度明显增加,如果抑制胶质瘤组织中的棕榈酰化修饰水平,将会激活胶质瘤细胞凋亡信号通路,并且引发内质网应激反应,这为探索棕榈酰化与胶质瘤的关系提供了重要依据。八聚体结合转录因子4(octamer-binding transcription factor 4,OCT4)基因多在肿瘤组织和细胞系中表达,在肿瘤干细胞维持和自我更新过程中发挥重要作用。化疗耐药性是肿瘤干细胞的重要特征之一,研究表明,OCT4 表达水平与化疗耐药呈正相关,OCT4能够作为化疗耐药和预后预测标志物[23-24]。研究发现,棕榈酰化OCT4 易与性别决定区Y 框蛋白4(sex determining region Y-box 4,SOX4)结合,共同维持胶质瘤干细胞持续增殖,促进胶质瘤的发生发展[25]。目前已经证实巴豆酰化修饰、棕榈酰化修饰和琥珀酰化修饰方式可能与胶质瘤的进展有关,并且为进一步开发修饰相关的肿瘤治疗药物提供了可能,然而这些修饰方式对胶质瘤耐药的调控尚未见报道,还有待于进一步的研究加以证实。
1.3 非编码RNA 调控
1.3.1 长链非编码RNA(long non-coding RNA,lncRNA)对胶质瘤耐药的调控 lncRNA 是长度>200 个核苷酸的异质RNA,可以影响细胞增殖、分化、凋亡,在诱导肿瘤的发生、发展、侵袭、迁移等生物学行为方面发挥着重要作用。lncRNA 没有编码蛋白质的能力,但是可以调节基因的表达。其中,肺腺癌转移相关转录本1(metastasis associated lung adenocarcinoma transcript 1,MALAT1)和H19在胶质瘤细胞对TMZ 耐药中的作用得到了充分研究。MALAT1 又称核富集丰富转录本2(nuclear enriched abundant transcript 2,NEAT2),是胶质瘤组织中高表达的一种致癌lncRNA。研究表明,在TMZ 耐药GBM 患者的GBM 组织中,MALAT1 表达显著上调,其通过抑制微小RNA(microRNA,miRNA)-203 的表达,导致GBM 对多种药物产生耐药性[25]。Li 等[26]研究表明,下调MALAT1 的表达可抑制多药耐药蛋白1(multidrug resistance protein 1,MDR1)和多药耐药相关蛋白5(multidrug resistance association protein 5,MPR5)等多药耐药相关基因的表达,从而达到使胶质瘤细胞对TMZ 增敏的目的。MALAT1 过表达可通过上调锌指E 盒结合同源框1(zinc finger E-box binding homeobox 1,ZEB1)等E-钙黏蛋白(E-cadherin)的表达,促进上皮-间充质转化(epithelial-mesenchymal transition,EMT),提高胶质瘤的耐药性。另一项研究表明,MALAT1 表达下调可导致耐药的胶质瘤细胞中miRNA-101 表达上调,糖原合成酶激酶(glycogen synthase kinase,GSK)-3β表达下调,克服胶质瘤细胞对TMZ 耐药[27]。H19 是另一种常见的致癌lncRNA,常在胶质瘤组织和细胞中过表达,尤其在TMZ 耐药的胶质瘤U251 和M059J 细胞中表达高度上调[28]。H19 可使细胞周期蛋白(cyclin)D1 基因表达上调,影响细胞周期转变,促进胶质瘤细胞增殖,并防止胶质瘤细胞周期停留在G2/M 期,从而抑制TMZ 诱导的细胞凋亡[3]。抑制H19 的表达可使耐药基因MDR、MRP、三磷酸腺苷结合盒亚家族G成员2(adenosine triphosphate binding cassette subfamily G member 2,ABCG2)表达明显降低,也可通过上调E-cadherin 的表达,降低波形蛋白(vimentin)和ZEB1 的表达水平,从而抑制EMT 过程,使胶质瘤细胞对TMZ 更加敏感[29]。除上述两种lncRNA 外,还有一系列其他lncRNA 以多种不同途径参与调控胶质瘤耐药[30-46](表1),这些lncRNA 可以作为克服TMZ 耐药性的有希望的治疗靶点,从而提高TMZ 化疗的临床疗效。
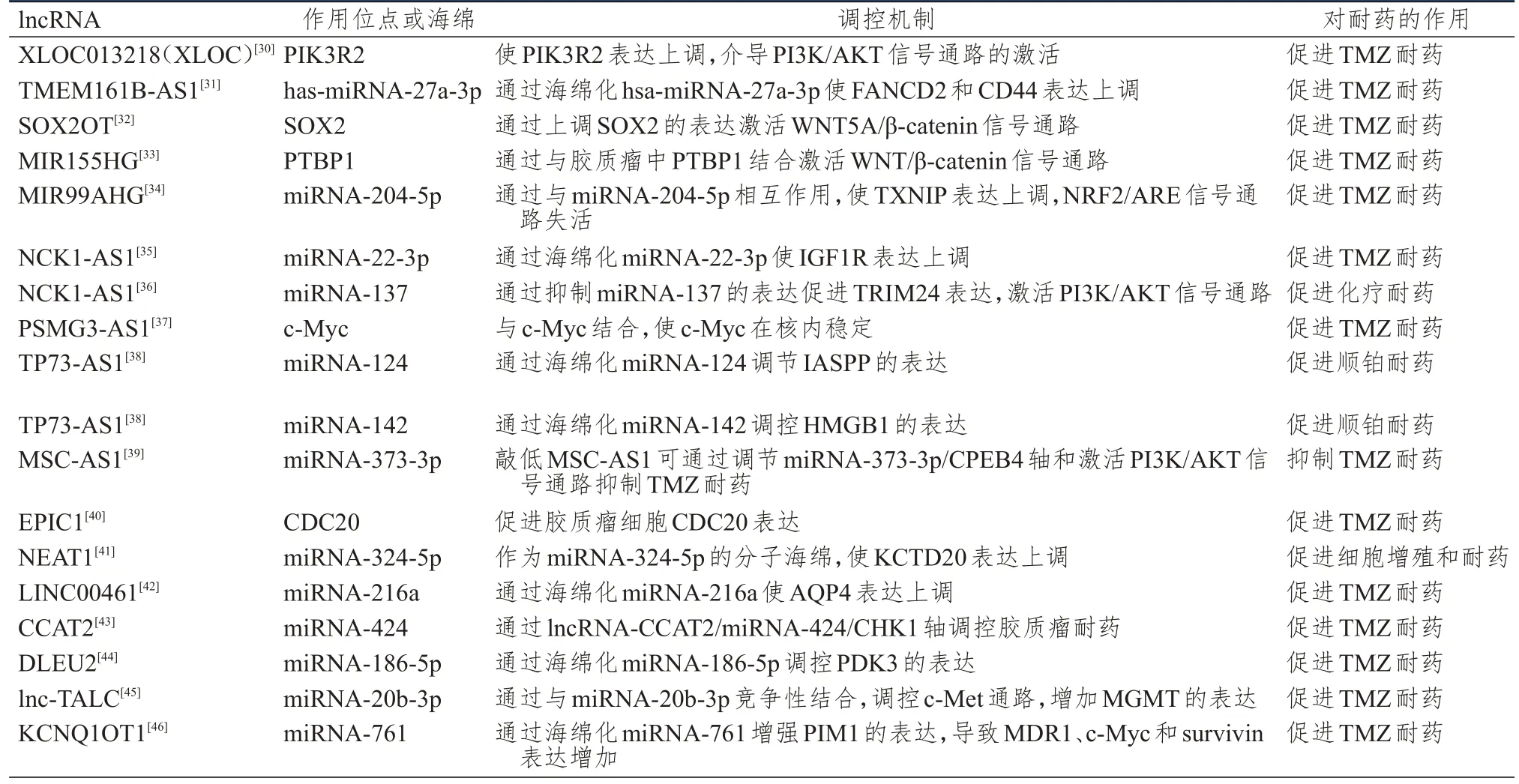
表1 lncRNA 对胶质瘤耐药的调控
1.3.2 miRNA 对胶质瘤耐药的调控 基因翻译水平可影响各种细胞进程[47]。单个miRNA 可以同时影响GBM 中多个mRNA,而GBM 的单个mRNA也可被一个或多个miRNA 调节[48]。近年来的研究表明,miRNA 异常表达与胶质瘤耐药密切相关,且其调控机制错综复杂,常见的调控方式主要包括介导细胞凋亡、MGMT 对DNA 损伤的修复以及EMT 等[49]。例如,miRNA-221 通过使其靶点磷酸酶张力蛋白同源物(phosphatase and tensin homolog,PTEN)表达下调,激活磷脂酰肌醇-3-羟激酶(phosphatidylinositol 3-hydroxy kinase,PI3K)/蛋白激酶B(protein kinase B,PKB,又称AKT)信号通路,并调控EMT 相关基因的表达,促进胶质瘤耐药[50]。研究表明,靶向动力蛋白3(dynamin 3,DNM3)可诱导胶质瘤发生发展及对TMZ 耐药[51]。Areeb 等[52]研究表明,miRNA-221 还可以通过下调表皮生长因子受体(epidermal growth factor receptor,EGFR)的表达增强胶质瘤对TMZ 和放疗的耐受性。其他多种miRNA 也可通过靶向不同的mRNA 以多种不同途径参与胶质瘤耐药[53-62](表2)。miRNA 表达与胶质瘤化疗耐药密切相关,目前的研究表明,利用miRNA 抑制剂或类似物降低因miRNA 表达失调引起的耐药,有望成为逆转胶质瘤耐药的有效方法。

表2 miRNA 对胶质瘤耐药的调控
1.4 染色质重塑
染色质重塑是由三磷酸腺苷(adenosine triphosphate,ATP)供给能量,在染色质重塑复合体的介导下,使得核小体移动或解离的过程。染色质重塑复合体具有ATP 酶活性,根据发挥催化作用的ATP 酶亚基的不同,可以将复合体分为4 类:交配型转换(mating type switching,SWI)/蔗糖不发酵(sucrose non-fermenting,SNF)复合体、模拟开关(imitation switch,ISWI)、染色质域-解旋酶-DNA 结合蛋白(chromodomain helicase DNA binding protein,CHD)、INO80。这些复合体及其相关蛋白与细胞周期的激活和抑制、DNA 修复、DNA 甲基化及DNA 转录有关[12]。染色质重塑复合体关键基因的突变可使核小体无法定位,导致染色质重塑失败,基因表达异常,最终可能导致肿瘤的发生发展及耐药的产生[47]。其中,SWI/SNF 复合体得到了较为充分的研究,SWI/SNF 复合体的Brahma 相关基因1(Brahma-related gene 1,BRG1)催化亚基可促进GBM 细胞的恶性表型及对TMZ 耐药。研究显示,敲除BRG1增加了GBM 细胞对TMZ 和顺铂的敏感性,BRG1 表达下调也增加了GBM 干细胞在体外对TMZ 的敏感性。含BRG1 的SWI/SNF 复合体可调控多种DNA 修复途径,包括通过同源重组修复DNA 双链断裂[63]。Ganguly 等[64]敲低胶质瘤起始细胞(glioma-initiating cell,GIC)X16 中的BRG1,并采用TMZ 处理,结果发现,敲低BRG1后,GIC X16 中MGMT 表达降低,且对TMZ 的敏感性显著提高。由于染色质结构的复杂性,靶向染色质重塑研究GBM 的化疗耐药是一项艰巨的挑战,目前的研究集中在利用抗染色质重塑剂防止动态染色质重排来克服GBM 的耐药性。
2 逆转胶质瘤耐药的表观遗传学方法及前景
随着对表观遗传学与胶质瘤耐药机制关系研究的不断深入,从表观遗传的角度治疗胶质瘤,逆转胶质瘤耐药的前景变得更为明朗。如前所述,MGMT 表达上调可促进胶质瘤对TMZ 耐药,抑制MGMT 的表达为逆转TMZ 耐药提供了一种可能。β干扰素可通过下调MGMT 的表达抑制胶质瘤细胞对TMZ 耐药,与TMZ 联合治疗胶质瘤为未来的临床应用提供了广阔的前景[65]。一氧化氮供体S-亚硝基-N-乙酰青霉胺(S-nitroso-N-acetyl penicillamine,SNAP)也可通过下调MGMT 的表达抑制TMZ 耐药[66]。另有研究表明,白藜芦醇可通过抑制WNT 信号通路激活、下调MGMT 的表达增加胶质瘤细胞对TMZ 的敏感性[67]。高选择性磷酸甘油酸脱氢酶抑制剂NCT503 能够抑制丝氨酸合成途径,可能通过干扰WNT/β-联蛋白(β-catenin)通路和活性氧介导的DNA 损伤,降低MGMT 的表达。因此,NCT503 和TMZ 联合治疗可能是一种新的治疗策略[68]。小分子抑制剂EPIC-0412 可通过影响p21-E2F 转录因子1(E2F transcription factor 1,E2F1)轴抑制GBM 细胞的DNA 损伤修复,还可以通过与MGMT启动子区域的激活转录因子3(activating transcription factor 3,ATF3)-p-p65-HDAC1轴相互作用使MGMT基因沉默,以增强胶质瘤细胞对TMZ 的敏感性[69]。在胶质瘤中,一些特殊的HDAC 也可促进胶质瘤耐药。Yelton 和Ray[70]的研究显示,HDAC 抑制剂可引起组蛋白或非组蛋白乙酰化增加,使胶质瘤细胞染色质构象更为开放,更易于TMZ 破坏DNA,这也提供了一种绕开TMZ 耐药治疗胶质瘤的方法。妥巴他汀A 是一种HDAC抑制剂,可以逆转胶质瘤EMT 进程,进而增强TMZ 诱导的细胞凋亡[71]。NBM-BMX 是一种HDAC8 抑制剂,可通过下调β-catenin/c-Myc/SOX2信号通路促进细胞凋亡,这种抑制剂也可以抑制MGMT 的表达,使胶质瘤细胞对TMZ 增敏[72]。下调SWI/SNF 复合体的BRG1 催化亚基的表达也为逆转胶质瘤耐药提供了一种可能,BRG1 溴结构域抑制剂PFI-3 增强了TMZ 在GBM 细胞中的抗肿瘤活性,也可达到克服耐药的目的[73]。研究表明,仿生纳米声敏剂系统与无创超声驱动相结合,可有效穿过血脑屏障,靶向递送化疗药物,产生的活性氧不仅可以诱导胶质瘤细胞凋亡,还可以下调耐药相关因子的表达,增强化疗敏感性[74]。另外,一系列非编码RNA 也为逆转胶质瘤耐药提供了可能的位点,这有待于进一步的研究。
3 小结与展望
近年来,研究者对表观遗传学在胶质瘤耐药中调控机制的不断深入研究,为未来胶质瘤的治疗提供了新的方向,为逆转胶质瘤耐药提供了新的可能。MGMT、HDAC等基因表达上调、SWI/SNF复合体的BRG1 催化亚基以及一系列非编码RNA都可以促进胶质瘤耐药,同时也为逆转胶质瘤耐药提供了可能。MGMT、BRG1 以及一些特定的HDAC 抑制剂对逆转胶质瘤耐药、提高治愈率具有重要作用。TMZ 与抑制剂联合治疗可更好地发挥TMZ 的疗效,达到治疗胶质瘤的目的。然而胶质瘤的耐药机制错综复杂,在表观遗传学的影响下胶质瘤的发展及预后更加难以预测,未发现的耐药机制以及各种机制间的补偿作用也导致目前解决胶质瘤耐药的效果不是很理想。因此今后的研究可以着手于目前尚不明确的耐药机制以及耐药机制间的补偿作用,为逆转胶质瘤耐药提供更多可能,并开发更多可逆转胶质瘤耐药的药物或其他化疗药物。希望未来能够早日突破胶质瘤耐药难题,改善患者预后。
猜你喜欢
畜牧兽医学报(2022年3期)2022-03-30
中国畜牧兽医(2022年1期)2022-02-15
河北果树(2021年4期)2021-12-02
上海公路(2019年3期)2019-11-25
现代泌尿外科杂志(2019年10期)2019-10-31
生物学通报(2019年2期)2019-06-15
福建基础教育研究(2019年10期)2019-05-28
华东理工大学学报(自然科学版)(2015年4期)2015-12-01
医学研究杂志(2015年9期)2015-07-01
云南中医学院学报(2014年2期)2014-11-07
