
微波辅助萃取-液质联用法快速检测食品种九种维生素
2023-12-14 06:03鲁长海何晓峰刘云苑陈洁琼
广州化工 2023年14期
鲁长海,何晓峰,林 绪,刘云苑,陈洁琼
(广电计量检测集团股份有限公司,广东 广州 511450)
维生素是人类为维持正常生理机能不可缺少的一类微量有机化合物,多为酸、醇和酚类物质。维生素的种类很多,根据其溶解性可分为水溶性维生素和脂溶性维生素两大类。水溶性维生素主要包括维生素C(又叫抗坏血酸)及B族维生素。其中B族维生素有12种以上,被世界一致公认的有9种[1-9]。维生素B族包括维生素B1(硫胺素)、维生素B2(核黄素)、维生素B3(烟酸、烟酰胺)、维生素B5(泛酸钙)、维生素B6(吡哆醇、吡哆醛、吡哆胺)、维生素B12(钴胺素)、维生素B9(叶酸)、维生素B7(生物素)等,对保健食品的质量监控、疾病的诊断和治疗以及生命科学等领域也具有重要的意义[10-11]。
现有维生素方法对于复杂基质样品检测前处理繁琐、速度慢,准确度不高且大多面对复杂基质中的各种维生素分离不佳,本问提供了一种快速检测方法,使用微波辅助萃取大幅缩短提取时间,使用液质联仪同时检测食品中9种维生素:硫胺素(维生素B1)、核黄素(维生素B2)、烟酰胺(维生素B3)、泛酸(维生素B5)、吡哆醇(维生素B6)、生物素(维生素H)、叶酸(维生素B9)、维生素B12和维生素C。
1 实 验
1.1 材料与试剂
标准物质:盐酸硫胺素(94.43%),Dr;核黄素(97.6%)、盐酸吡哆醇(99.9%)、D-生物素(99.9%)、L-抗坏血酸(99.7%),安谱;烟酰胺(99.9%)、维生素B12(96.5%)、叶酸(89.9%),BePure;D-泛酸钙(99.9%),天津阿尔塔。
试剂:盐酸(优级纯),国药集团化学试剂有限公司;氨水(化学纯),广州化学试剂厂;乙醇(色谱纯),CNW;甲醇(质谱级),Sigma;甲酸(质谱级),CNW;乙酸铵(质谱级),LiChropur;偏磷酸,国药集团化学试剂有限公司。
耗材:HLB固相萃取小柱(500 mg,6 mL),Biocomma;有机系滤膜(津腾,尼龙66,13 mm,0.22 μm)。
耗材:市售某品牌多维片。
1.2 仪器与设备
AB 4500超高效液相色谱-质谱/质谱联用仪,美国SCIEX公司;超声波清洗器,天津奥特赛恩斯仪器有限公司;UV-2600i紫外可见分光光度计,日本岛津;XS205DU电子分析天平(0.001 mg),瑞士梅特勒;ME204E电子分析天平(0.01 mg),瑞士梅特勒;Milli-Q Advantage超纯水机,美国密理博;高速离心机,蜀科;涡旋振荡器,美国SI。
1.3 实验方法
1.3.1 标准溶液配制
(1)标准储备液的配制
标准物质称量前须经五氧化二磷干燥24 h,称取盐酸硫胺素、盐酸吡哆醇、泛酸、生物素、烟酰胺、维生素B12各约10 mg,分别置于10 mL容量瓶中,用20%甲醇水溶液(用磷酸调节pH值为4.0)定容至刻度,作为储备液,浓度约为1 000 μg/mL;称取核黄素约10 mg,分别置50 mL棕色容量瓶中,用5 mL盐酸(1+1)超声溶解后20%甲醇水溶液(用磷酸调节pH值为4.0)定容,浓度约为200 μg/mL;称取叶酸约10 mg,置10 mL容量瓶中,加入约1 mL水,加入1滴氨水,溶解后用20%甲醇水溶液(用磷酸调节pH值为4.0)定容,浓度约为1 000 μg/mL。
(2)混合标准中间液的配制
配制标准中间液前,核黄素按照GB 5009.85附录A进行浓度校准,烟酰胺按照GB 5009.89附录B进行浓度校准,盐酸吡哆醇按照GB 5009.154附录A进行浓度校准。
准确吸取各维生素标准储备液适量,用水稀释至最终浓度分别为1.00 μg/mL:盐酸硫胺素、核黄素、盐酸吡哆醇、生物素、维生素B12;2 μg/mL:烟酰胺、泛酸、叶酸;5 mL;20 μg/mL:L-抗坏血酸。
(3)标准系列工作液的配制
取不含维生素的空白样品按照样品处理方法制得空白基质溶液。分别吸取混合标准中间液0.10 mL、0.20 mL、0.40 mL、0.80 mL、1.00 mL、2.00 mL、4.0 mL、5.0 mL于10 mL棕色容量瓶中,用空白基质溶液定容至刻度混匀。此标准混合系列工作液中盐酸硫胺素、核黄素、盐酸吡哆醇、生物素、维生素B12的浓度分别为10 ng/mL、20 ng/mL、40 ng/mL、80 ng/mL、100 ng/mL、200 ng/mL、400 ng/mL、500 ng/mL;烟酰胺、泛酸、叶酸的浓度分别为20 ng/mL、40 ng/mL、80 ng/mL、160 ng/mL、200 ng/mL、400 ng/mL、800 ng/mL、1 000 ng/mL;L-抗坏血酸浓度分别为200 ng/mL、400 ng/mL、800 ng/mL、 1 600 ng/mL、2 000 ng/mL、4 000 ng/mL、8 000 ng/mL、10 000 ng/mL。
1.3.2 样品前处理
(1)提取
准确称取混合均匀的试样1 g(精确至0.001 g)于 50 mL棕色容量瓶中,加入40 mL 20%甲醇水溶液(用磷酸调节pH值为4.0),摇匀,置微波萃取仪480 W萃取30 s,取出冷却后加水溶液至刻度,混匀。以10 000 r/min离心5 min。取上清液待用。
取上清液1 mL至100 mL棕色容量瓶中,加水稀释至刻度,混匀。稀释液过0.22 μm 水相滤膜过滤后,供液相色谱-串联质谱仪测定。
1.3.3 液相色谱-串联质谱条件
(1)液相色谱条件
采用Waters ACQUITY UPLC®HSS T3色谱柱(2.1 mm×100 mm×1.8 μm);进样量5 μL;流速:0.3 mL/min;柱温40 ℃;梯度洗脱:流动相A为20 mmol/L乙酸铵含0.1%(V/V)甲酸水,流动相B为0.1%甲酸甲醇,洗脱程序见表1。
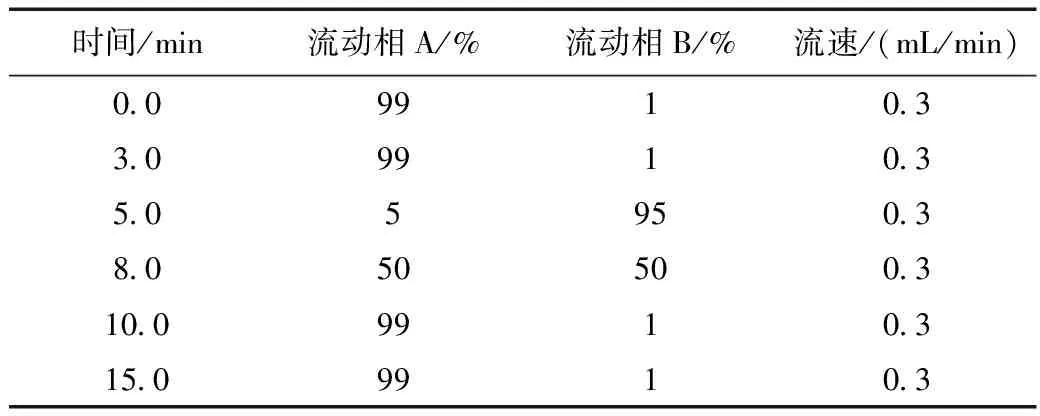
表1 流动相梯度洗脱条件
(2)质谱条件
气帘气流速:30 L/min;雾化气流速(GS1):50 L/min;辅助加热气流速(GS2):50 L/min;碰撞气(CAD):中等强度(medium);辅助加热气温度:550 ℃;喷雾电压:5 500 V(ESI+);扫描模式:多反应监测模式(MRM)。
1.3.4 定性测定
(1)采用优化后的仪器方法及质谱参数对样品进行扫描,如目标物无响应则样品未检出,无需定量。
(2)如扫描后出现目标物阳性则按照超高效液相色谱-三重四极杆串联质谱条件测定试样和标准工作溶液,记录试样和标准溶液中各化合物的色谱保留时间,当试样中检出与某标准品色谱峰保留时间一致的色谱峰(变化范围在±2.5%之内),并且试样色谱图中所选择的监测离子对的相对丰度比与相当浓度标准溶液的离子相对丰度比(k)的偏差不超过表2规定的范围,可以确定试样中检出相应化合物。

表2 定性确证时相对离子丰度的最大允许偏差
1.3.5 定量测定
将标准工作溶液(1.3.4)按仪器参考条件进行测定,得到相应的标准溶液的色谱峰面积。以标准工作溶液的浓度为横坐标,以定量离子的色谱峰的峰面积为纵坐标,采用线性过原点绘制曲线,外标法。
1.3.6 结果计算
如目标物检出按下列公式(1)计算出样品中目标物的含量:
(1)
式中:X——试样中各待测物的含量,mg/kg
c——待测物的浓度,μg/L
V——样液最终定容体积,mL
m——试样溶液所代表的质量,g
f——稀释倍数
计算结果应扣除空白值,保留三位有效数字。
2 讨论与结论
2.1 色谱与质谱条件优化
采用标准储备液直接作为质谱参数优化用溶液,采用针泵流动注射连续进样的方式进行质谱全扫描检测,得到被测化合物一级质谱图,找出两者对应的母离子,再进行二级质谱扫描,得到相应的子离子信息。然后优化两者的碰撞能量及去簇电压等参数,使两者的母离子与特征碎片离子强度最佳,各标准品的定性和定量离子对以及最优质谱参数见表2。各维生素物的质谱检测参数见表3。

表3 目标化合物的质谱检测参数
2.2 微波辅助萃取溶剂的优化
水是水溶性维生素的最优提取溶剂,甲醇和乙腈是常用提取溶剂,分别选择水溶液(磷酸调节pH=4.5)、20%甲醇水溶液(用磷酸调节pH值为4.0)、20%乙腈水溶液(用磷酸调节pH值为4.0)作为提取溶剂,分别对同一某品牌多维片进行测定,结果回收率分布如表4所示。

表4 不同提取溶剂回收率分布图
经过比较,甲醇水溶液的回收率明显更为优异,因此选择甲醇水作为提取溶剂。
2.3 提取液pH的优化
另行将提取液调节pH为10.0和4.0进行检测,结果除碱性环境中叶酸回收率较大提高至92%,其余品种维生素回收率均大幅下降,因而最终仍然以pH=4.0的甲醇水作为提取溶剂。
2.4 萃取时间的优化
将微波的时间分别调整为10 s、20 s、30 s、40 s、50 s,然后按照流程测试,结果如表5所示。

表5 不同萃取时间条件下的回收率
由表5可以看出,随着时间的增加,在10~30 s的时间内,维生素的回收率逐渐增加,30~50 s,维生素的回收率趋于平稳,甚至略有下降,分析原因,可能是微波有助于维生素的溶出,30 s时达到最佳提取效率,30 s后部分维生素略有分解,因此,最终选择萃取时间为30 s。
2.5 微波功率的优化
调节微波萃取的功率分别为160 W、320 W、480 W、640 W、800 W,分别对某品牌多维片样品进行微波辅助萃取,维生素的回收率如表6所示。

表6 不同微波功率对样品回收率的影响
由表6可知,微波功率对提取效率有较大的影响,当功率为480 W时,达到最大提取效率,有少许维生素降解的迹象,超过480 W后存在较为明显的降解,所以选择微波萃取的功率为480 W。
2.6 溶剂溶剂用量的优化
选用磷酸调节pH=4.0的甲醇水溶液作为提取溶剂,选择微波功率480 W,萃取时间30 s,30 mL、40 mL、50 mL、60 mL、70 mL提取液进行微波萃取,萃取的各种维生素回收率如表7所示。

表7 不同体积提取溶剂用量对应的维生素回收率

表8 方法特异性实验结果
从表7可以看出,随着提取溶剂体积的增加提取回收率逐渐上升,当提取溶剂用量到达50 mL后维生素的回收率到达平衡点,再增加提取溶剂用量对结果影响不大,在此条件下的回收率已能满足《GB 5009.295-2023 食品安全国家标准 化学分析方法验证通则》的要求。
2.7 方法特异性
分别采用阴性基质以及向阴性基质中添加盐酸硫胺素、核黄素、烟酰胺、泛酸、盐酸吡哆醇、生物素、叶酸、维生素B12和L-抗坏血酸标准溶液进行专属性测定,发现阴性基质中无对检测造成干扰的杂质。
2.8 检测限和定量限
取制备的空白基质溶液,加入适量维生素B1、B2、烟酰胺、泛酸、吡哆醇、生物素、叶酸、B12和L-抗坏血酸标准储备液(维生素B12所用空白溶液为水)。逐级稀释,进入仪器分析,以大于3倍信噪比时的溶液浓度为检出限,大于10倍信噪比时的溶液浓度为定量限。
2.8.1 检测限
维生素B1、维生素B2、烟酰胺、泛酸、吡哆醇、叶酸、生物素、维生素B12的检测限均为1.0 ng/mL,当称样量为1 g,最终定容体积为50 mL时,维生素B1、B2、烟酰胺、泛酸、吡哆醇、叶酸的、生物素、维生素B12的检测限均为0.05 μg/g;维生素C(L-抗坏血酸)的检测限为10.0 ng/mL,当称样量为1 g,最终定容体积为50 mL时,维生素C的检测限为0.5 μg/g。
2.8.2 定量限
维生素B1、B2、烟酰胺、泛酸、吡哆醇、叶酸、生物素、维生素B12的定量限均为3.0 ng/mL,当称样量为1 g,最终定容体积为50 mL时,维生素B1、B2、烟酰胺、泛酸、吡哆醇、叶酸、生物素、维生素B12的定量限均为0.15 μg/g;维生素C(L-抗坏血酸)的检测限为30.0 ng/mL,当称样量为1 g,最终定容体积为50 mL时,维生素C的定量限为1.5 μg/g。
2.9 测定范围
按照1.3.1配制一系列的标准工作溶液进行测试,然后将结果以各组分定量离子的峰面积为纵坐标,各组分含量为横坐标对其进行线性回归,的到各组分的线性回归方程和相关系数R2,结果见表5,线性关系图见图3。表明以该方法测试维生素B1、B2、烟酰胺、泛酸、吡哆醇、生物素、叶酸、B12和L-抗坏血酸的含量,在表9中所列线性范围内,方法线性关系良好。

表9 测定范围测试结果
2.10 精密度试验
为保证方法的重现性,以某品牌多维片为样品进行了精密度试验。平行配制六份样品样液进行测试,测试结果如表10所示。六份样品测试结果中9种维生素的结果之间的RSD均小于5%,精密度良好。

表10 方法精密度测试结果

表11 方法正确度试验结果
2.11 正确度试验
按照1倍定量限浓度、2倍定量限浓度、10倍定量限浓度进行加标,每个浓度平行测定三次,最终所有回收率均在80%~110%范围内。
3 结 论
本文建立了一种针对食品中9种维生素的利用微波辅助萃取快速提取,使用超高效液相色谱串联质谱法同时快速检测的方法。本文研究了微波辅助萃取食品中维生素的溶剂组成以及萃取液的pH值、微波萃取的功率以及时间等,最终得到了微波辅助萃取的最佳条件为:使用pH=4的甲醇水作为萃取溶剂,在480 W的功率下微波萃取30 s。在此条件下,该方法中所有9种维生素成分均在10~200 ng/mL范围内线性良好,相关系数R2均大于0.999,方法检出限均低于1.0 ng/mL,定量限均低于3.0 ng/mL,回收率均在80%~110%之间,连续六次平行实验结果之间的相对标准偏差均<10%。相比较现有技术,该方法前处理时间大幅缩短,可极大提高分析效率,同时,方法灵敏度高,选择性好,结果准确可靠,可应用于食品中多种维生素的同时快速检测。
猜你喜欢
兽医导刊(2020年23期)2020-12-30
饮食与健康·下旬刊(2019年4期)2019-03-09
发酵科技通讯(2017年3期)2017-09-12
农村青少年科学探究(2017年6期)2017-09-11
——水溶性维生素泛酸篇
质量与标准化(2016年5期)2016-04-12
合成化学(2015年4期)2016-01-17
饲料工业(2016年23期)2016-01-09
西华师范大学学报(自然科学版)(2015年3期)2015-02-27
药学进展(2014年9期)2014-03-08
药学与临床研究(2014年3期)2014-03-06
