
三元硝酸熔盐体系的相图研究
2023-12-14 06:03朱文龙
广州化工 2023年14期
朱文龙
(北京博鼎诚工程设计有限公司广州分公司,广东 广州 510655)
熔盐具有导热系数大、热稳定性好、热容量大等突出优势,是高温蓄热材料的最佳选择之一,广泛应用于太阳能热发电等新能源领域。太阳能热发电的开发利用和工业系统节能均取决于储热技术的发展状况,研究开发高效能低成本熔盐蓄热传热材料,可在一定程度上解决制取新能源所面临的高成本、低产率等问题。良好的蓄热传热材料是蓄热系统运行的重要环节,更是缓解当下全球能源危机的关键所在。
相图是在材料科学中应用广泛,是材料发展和生产的重要基础,对材料科学的进步起关键性作用,因此,相图工作与工业生产是密切相关的。目前已经测得的熔盐相图数达几千张,主要是三元或三元以下体系的相图。进行具体物质体系相图计算时需要设计具体算法才能较为准确地得到该体系相图。
本文从材料科学发展和工业生产对相图的急切需求的工程背景,提炼开发出高温蓄热技术所需要解决的低成本高性能熔盐材料体系构建的关键科学问题,作为研究目标。发展传热蓄热材料性能可控的制备新技术,获得对高效储能技术创新发展有导引与基础支撑作用的科学认知积累。
本文主要涉及三元硝酸熔盐体系LiNO3- KNO3-Ca(NO3)2的相图计算,其是从纯熔盐热力学数据出发,基于化学势相等和吉布斯自由能最小原理,计算二元体系的共熔点和相互作用系数,在二元体系基础上计算三元体系的共熔点及相应的组成。
1 相图计算
1.1 相图计算的基本原理和方法
能量最小化原则是相图计算的基本原理,即在给定的封闭环境中,当整个体系达到热力学平衡时,整体总能量应取极小值。常用的是吉布斯自由能最小法和等化学势方法[1-2]。
(1)吉布斯自由能最小法当整个体系达到热力学平衡后,封闭体系中总吉布斯自由能G应取极小值。
G=Gmin
(1)
吉布斯自由能最小法又称寻优法,即对于给定的体系,当温度和总成分一定时,通过对整个体系总吉布斯自由能求最小值,得到平衡态时整个体系中各分相的具体组成。
(2)等化学势法当整个体系处于热力学平衡状态时,任一组元i在各相中的化学势相等,从而得到一组方程:
(2)
也可以采用某一组分的偏摩尔自由能表示为:
(3)
式中:P为体系的总相数;i为其中某一组分;C为体系的组元数。
利用具体的数值计算方法,确定上述的多项非线性方程组的解,即可得达到平衡态时整个体系中各个相的具体组成和温度。
1.2 溶液热力学模型
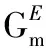
(1)理想溶液模型
在理想溶液模型中,主要假设的是溶液中各组分间的相互作用力很小,近似忽略不计,故溶液中过剩摩尔吉布斯自由能为零。在实际应用过程中,理想溶液不考虑原子间的相互作用力的假设是很不准确的,如固熔体中各组分间的相互作用力就远比液态中各组元间的相互作用大,但是在整个体系热力学实验数据较少的状况时,其液态热力学性质可近似用理想溶液模型代替。
(2)正规溶液模型
1929年,Hildebrand[3]提出了正规溶液模型,该溶液模型针对前面提到的固熔体体系的缺陷,主要考虑了每一个原子附近都会有离其最近邻原子,但是这个原子又与中心原子种类无关,而且这个原子在体系中完全是非有序排列,故此时体系的过剩熵可近似为零。溶液模型热力学性质如下:
SE=0
(4)
(5)
(6)
式中:C为溶液的总组元数;λij为二元溶液体系i-j间的相互作用系数,qi为离子电荷数,xi为i组分的摩尔分数。
对于正规溶液而言,其相互作用系数λij假设的是与温度、组成无关,可以近似为零级处理,故只能用来计算体系间相互作用系数不大的溶液的热力学性质。
(3)共形离子溶液模型
共形离子溶液模型(CIS)是以统计力学微扰理论为基础,其是将实际溶液看作为理想溶液的微扰体系,通过统计力学微扰理论来计算溶液体系的热力学函数。这个溶液理论不仅考虑了溶液中离子间近程库仑力的作用,还考虑了远程库仑力的影响。考虑的因素比较简单,故比较适合于阳离子(或阴离子)半径相差较大且热力学数据缺乏的多元熔盐体系。
在以上描述的模型中,理想溶液模型没有考虑周围原子的影响,正规溶液模型只考虑了最邻近原子的影响,两者都不准确;共形离子溶液模型考虑了溶液中离子间近程库仑力的作用和远程库仑力的影响,这个模型考虑的因素不是很复杂,比较适合用于缺少实验数据的多元熔盐体系的理论计算。故本文将采用共形离子溶液模型(CIS)对三元熔盐体系进行理论研究。
2 二元熔盐体系相图计算
2.1 计算原理
二元熔盐体系相图计算主要利用的是“硬球”离子相互作用模型[4-6]忽略二元物质混合过程中阳离子的非随机分布、体积变化和离子极化等因素的影响,AX-BX二元熔盐体系的混合能ΔEm如下:
(7)

(8)
式中:U为固态标准晶格能,可根据Kapustinskii公式得到:
(9)
式中:ν为一分子物质含有的离子数;r-、r+为阴阳离子半径;Z-、Z+为阴阳离子电数。
忽略二元物质混合时体积变化,过剩混合焓与体系混合能相等,即
ΔHE=ΔEm
(10)
“硬球”模型忽略二元物质混合过程中阳离子的非随机分布,则过剩混合熵ΔSE=0,故:
(11)
考虑二元熔盐体系按正规溶液模型混合,由式(5)在二元熔盐体系中的应用,可得二元熔盐体系过剩混合吉布斯自由能为:
(12)
结合式(7)、(11)、(12)知,相互作用系数为:
(13)
(14)
式中:γi(i=A,B)为各组分的活度系数;ne=qAnA+qBnB,由式(12)、(14)得,
RTlnγi=qi(1-Xi)2λ
(15)
固态组分A与A、B二组分液态混合物在温度TA条件下达到平衡时,其液态混合物中i(i=A,B)组分的活度αi可以通过下面的公式计算:
(16)
式中:ΔHf(i)为组分i的熔化焓,可以通过以下公式计算:
(17)

联立式(15),(16),(17)得到组分i(i=A,B)平衡Ti-Xi的关系式如下:
(18)

2.2 基础数据
根据熔盐手册和文献[7-10]得到的各种熔盐的阴阳离子半径、熔点、相变焓和比热容等热物性参数分别列于表1和表2。

表1 熔盐离子半径

表2 熔盐物性参数
2.3 结果与讨论
基于式(18)及二元熔盐体系组分组成的归一化关系式,利用计算机编程可进行计算得出二元熔盐相图,如图1所示。

图1 二元熔盐计算相图与实验相图[11-13]
对比二元熔盐计算相图与实验相图可知,LiNO3-NaNO3,LiNO3-KNO3体系的计算相图结果与实验相图较为一致,LiNO3-Ca(NO3)2,KNO3-NaNO3,Ca(NO3)2-NaNO3,Ca(NO3)2-KNO3体系计算相图结果与实验相图相差很大。由表3可看出,LiNO3-NaNO3,LiNO3-KNO3体系共熔点计算值与实验值相差20 K以内,且共熔点处组成计算值与实验值接近,而LiNO3-Ca(NO3)2,KNO3-NaNO3,Ca(NO3)2-NaNO3,Ca(NO3)2-KNO3体系或共熔点计算值与实验值相差大,或共熔点处组成计算值与实验值相差大。

表3 二元熔盐体系的计算值与实验值
本文利用了正规溶液模型获取二元熔盐体系的相互作用系数λij,该模型假设λij与温度、组成无关,存在一定适用性,因此部分计算相图与实验相图相差较大可能是λij的准确性造成。为提高λij的准确性,本文将在计算三元相图时利用二元相图实验值反推得到λij,详见下文介绍。
3 三元熔盐相加体系相图计算
3.1 计算原理
对于三元体系AX-BX-CX,根据共形离子溶液理论,其过剩混合Helmholtz自由能ΔAE可表示为[14-16]:
(19)
式中:ΔAE为组分ij的过剩混合Helmholtz自由能。
当恒容恒压时,Helmholtz自由能A与吉布斯自由能G相差较小。故对于恒压凝聚态体系AX-BX-CX,其过剩混合吉布斯自由能ΔGE可表示为:
(20)
式中:ΔGE为组分ij的过剩混合吉布斯自由能。若以AX为组分1,BX为组分2,CX为组分3,则根据物质正规溶液模型,二元熔盐体系相互作用系数λ与温度、组成无关,其过剩混合吉布斯自由能ΔGE分别为:
(21)
(22)

RTlnγA=qAXB(XB+XC)λ12+qAXC(XB+XC)λ13-qAXBXCλ23
(23)
RTlnγB=qBXA(XA+XC)λ12-qBXAXCλ13+qBXC(XA+XC)λ23
(24)
RTlnγC=-qC(XAXB)λ12+qCXA(XA+XB)λ13+qCXB(XA+XB)λ23
(25)
联立式(23),(17),(18)得到组分A平衡TA-XA的关系式如下:

(26)
同理,得:

(27)

(28)
鉴于二元体系计算相图与实际相图存在较大偏差,现采用以下方法求解相互作用系数以提高准确性。
现由实验值反推相互作用系数λij,避免公式适用性问题,可提高数据置信度。若已知某二元熔盐体系共熔点、共熔点处体系组成及必要的熔盐基本物性参数,可以通过解式(18)及二元体系组分组成的归一化关系式组成的方程组,获得较为准确的相互作用系数。通常而言,二元熔盐相图的共熔点及共熔点处体系组成是容易获得的,故此法可行。
3.2 结果与讨论
基于式(26)至式(28)及三元熔盐体系组分组成的归一化关系式,利用计算机编程可进行计算得出三元熔盐相图如图2所示,将三元熔盐体系的共熔点列于表4中。

图2 三元熔盐体系Ca(NO3)2-KNO3-LiNO3计算相图(a)与

表4 三元熔盐体系的计算值与实验值
对比三元熔盐计算相图与实验相图,可知三元熔盐体系的计算相图结果与实验相图较为一致,体系共熔点计算值与实验值相差10 K以内,共熔点处组成计算值均与实验值较为吻合。
由此可见,以实验值为基础计算获得的相互作用系数可以提高相图计算的准确性,利用共形离子溶液模型计算三元熔盐体系可取得较好的效果。
4 结 论
本文在计算了二元熔盐体系的基础上进行了三元熔盐相加体系的相图计算,得出以下结论:
二元熔盐体系计算模型进行了简化且计算所用数据均是基础数据,而相图计算不能完全依靠理论获得,必须充分利用实验数据,因此造成二元熔盐体系中有部分体系的计算值与实验值相差较大。为此,在计算三元熔盐相加体系时,以二元熔盐体系共熔点及共熔点处体系组成为基础计算获得二元熔盐相互作用系数,提高了数据的置信度,在实际计算中发现三元熔盐体系计算相图与实验相图的重合性良好。
在二元熔盐相图计算基础上进行的三元熔盐相图计算结果表明,三元熔盐体系LiNO3-KNO3-Ca(NO3)2的共熔点为380.98 K,此时体系包含25.9mol% LiNO3,59.7mol% KNO3,14.4mol%Ca(NO3)2,与文献实验值相近。
猜你喜欢
氯碱工业(2021年5期)2021-09-10
陶瓷学报(2019年6期)2019-10-27
上海建材(2019年1期)2019-04-25
电线电缆(2017年2期)2017-07-25
材料科学与工程学报(2016年1期)2017-01-15
大学化学(2016年4期)2016-07-27
工业炉(2016年1期)2016-02-27
上海金属(2015年4期)2015-11-29
学园(2015年5期)2015-10-21
上海金属(2013年6期)2013-12-20
