
5-羟色胺选择性重摄取抑制剂对骨代谢的影响及机制Δ
2023-12-13 03:46周丽敏徐永祥东莞市人民医院药学部广东东莞523059
中国药房 2023年23期
周丽敏,徐永祥 (东莞市人民医院药学部,广东 东莞 523059)
5-羟色胺(5-hydroxytryptamine,5-HT)又称血清素(serotonin),是一种存在于中枢和外周的重要神经递质。其中,中枢5-HT在脑干中缝核的神经元合成;外周5-HT主要由肠嗜铬细胞合成,是体内大部分5-HT 的来源(95%),部分外周5-HT 还可来源于脂肪细胞、胰岛B 细胞、骨细胞[1―2]。5-HT 合成的主要限速酶是色氨酸羟化酶(tryptophan hydroxylase,TPH),其中TPH1 是肠嗜铬细胞、骨细胞合成5-HT 的限速酶,TPH2 则是中枢神经元、肠神经元合成5-HT的限速酶。由于5-HT无法穿透血脑屏障,因此中枢5-HT与外周5-HT相互独立。5-HT在胃肠道合成后可释放入血并贮存在血小板中,也可通过循环扩散到骨骼等组织[1―3]。5-HT 转运蛋白(serotonin transporter,SERT)是阻断5-HT 信号转导的必需分子,在神经元突触前膜、上皮细胞、血小板、骨细胞等均有表达[1]。当5-HT 被释放并介导受体作用后,其被SERT 再摄取从而被快速灭活[1]。在成骨细胞和破骨细胞中均存在5-HT受体和SERT,但不同骨细胞中分布的5-HT 受体类型不同[1,4]。既往研究显示,中枢和外周5-HT可能对骨形成产生相反的作用:肠源性5-HT与骨形成减少相关,而脑源性5-HT则有利于骨形成[2]。
5-羟色胺选择性重摄取抑制剂(serotonin-selective reuptake inhibitor,SSRI)是治疗抑郁症的一线药物,常用的有氟西汀、帕罗西汀、舍曲林、氟伏沙明、西酞普兰、艾司西酞普兰。SSRI 可通过拮抗SERT,抑制突触间隙5-HT 再摄取,提高5-HT 表达水平,从而改善抑郁症状[2]。除治疗抑郁症外,SSRI 也用于治疗焦虑症、睡眠障碍等其他疾病。但研究表明,SSRI可调节骨稳态——荟萃分析显示,SSRI 的使用与骨密度降低、骨折风险增加有关[5―6];另有研究显示,SSRI 会对骨重塑、胚胎骨骼发育产生不良影响[7―8];但也有一些研究显示SSRI 不影响骨代谢,甚至可对其产生积极影响[9―10]。SSRI 对骨代谢的影响与药物种类、药物浓度、治疗持续时间、骨骼部位、患者年龄等均存在关联性[11]。在该类药物中,氟西汀影响骨代谢的相关研究较多,而其他SSRI 类药物对骨代谢影响的相关研究则较少。目前服用SSRI的患者越来越多,且往往该类患者需要长期药物治疗,因此SSRI对骨代谢的影响不容忽视。本文就SSRI影响骨代谢的作用及潜在机制作一综述,为其临床合理使用提供参考。
1 SSRI对骨形成的影响及机制
成骨细胞在维持骨代谢稳态和骨再生过程中发挥着重要作用,可介导骨形成并调控破骨细胞的骨吸收。目前许多研究结果显示SSRI 可影响骨形成,其作用机制可能与以下几个途径有关。
1.1 影响成骨细胞增殖和成骨分化
有研究将接受10 mg/(kg·d)氟西汀治疗3周后的小鼠行骨折手术建立膜内成骨模型,结果显示氟西汀可导致骨缺损处细胞增殖减少、早期成骨分化标志物Runt相关转录因子2(Runt-related transcription factor 2,RUNX2)表达下降、矿物质沉积减少[12],提示SSRI 可通过抑制成骨分化影响骨折愈合。另一项研究的方法类似,用10 mg/(kg·d)氟西汀处理小鼠3 周后行骨折手术建立膜内成骨和软骨内成骨模型,在膜内成骨模型上观察到氟西汀可导致愈伤组织体积减小、成骨细胞数量和骨基质沉积减少;在软骨内成骨模型上也观察到氟西汀可导致愈伤组织体积减小、矿物质沉积率和骨形成率降低[13]。同时,在该两种模型上均显示出氟西汀联合β受体阻滞剂普萘洛尔后可以逆转对成骨分化的抑制。该研究还显示,氟西汀可降低骨髓间充质干细胞向成骨细胞分化的过程中碱性磷酸酶(alkaline phosphatase,ALP)的活性及成骨细胞特异性转录因子Osterix 的表达水平,减少矿化,但不能通过联合普萘洛尔逆转,提示氟西汀在体外对成骨分化具有直接抑制作用,普萘洛尔可能通过中枢介导逆转氟西汀的负面影响。另一些研究结果则不同,例如有研究在大鼠接受氟西汀10 mg/(kg·d)治疗16 周后分析其骨微结构,发现其股骨干骺端区域内单位骨面积的成骨细胞数量增加,提示氟西汀可增强成骨细胞增殖[9]。不同研究得出不同实验结果的原因可能是前两项研究为骨损伤模型而后一项研究并未作骨损伤处理。骨折后骨再生是一个复杂的过程,其愈合过程受多种因素(如血供、断端稳定性、炎症等)影响。膜内成骨和软骨内成骨是骨折愈合过程中的两种骨折修复形式,其中膜内成骨通常发生在骨折远端、近端或骨缺损(如胫骨、颅骨缺损)处,软骨内成骨则通常发生在长骨骨折(如股骨中段骨折)处[14]。由上述内容可知,氟西汀在骨损伤模型上表现出抑制成骨作用,而在未行骨损伤的模型上表现出促成骨作用,这提示氟西汀可对骨折愈合过程产生负面影响,但对原本未损伤的骨结构可能会有积极影响。此外,在其他SSRI 类药物中,Howie 等[15]的研究观察到经舍曲林处理3 d后,小鼠胚胎成骨细胞MC3T3-E1的增殖减少,而处理7 d 时的细胞增殖反而增加。在另一项体外研究中,研究者用西酞普兰处理人成骨肉瘤细胞Saos-2后,发现该药可促进成骨细胞增殖且不影响其矿化[16]。不同实验结果不仅与实验模型有关,还可能与不同细胞来源、药物品种、给药时间有关。由此可见,SSRI可通过影响成骨细胞的增殖和分化来影响骨形成。
1.2 增强肠道5-HT作用
环磷酸腺苷(cyclic adenosine monophosphate,cAMP)/蛋白激酶A(protein kinase A,PKA)/cAMP 反应元件结合蛋白(cyclic-AMP response binding protein,CREB)信号通路是促成骨分化和骨形成的重要信号通路。既往研究显示,肠道5-HT 与成骨细胞上的5-羟色胺1B 受体(5-hydroxytryptamine receptor 1B,Htr1B)结合,通过PKA 依赖途径抑制CREB 磷酸化,从而导致骨量减少[17]。实验显示,大鼠在灌胃氟西汀40 d后的骨形成标志物Ⅰ型前胶原氨基端原肽(procollagen type Ⅰ Nterminal propeptide,P1NP)的水平显著降低,而艾司西酞普兰未能显著影响P1NP 的表达;两药均能降低大鼠血清磷酸化CREB(phosphorylated CREB,pCREB)的水平,其中氟西汀组较艾司西酞普兰组更为显著[18]。可见,SSRI可能通过增强肠道5-HT的作用导致pCREB减少从而减少骨形成,且不同药物对骨形成的影响程度不同。另一项大鼠实验显示,使用可减少外周5-HT 的卡比多巴(脱羧酶抑制剂)后,可逆转艾司西酞普兰对骨形成标志物ALP 的负面影响[19],这再次验证了SSRI 可通过肠道5-HT介导骨形成减少。一项基于印度人群的横断面研究也得到了一致结论[20]——该研究发现,使用SSRI(平均持续时间11.84 个月)可导致患者血清P1NP水平显著降低,同样提示了SSRI 可减少骨形成;接受SSRI 治疗的患者血清中pCREB 的水平降低,而与脑源性5-HT 效应有关的核因子κB 受体活化因子配体(receptor activator of nuclear factor-κB ligand,RANKL)的水平则不受影响。综上所述,SSRI 可通过增强肠道5-HT途径介导骨形成减少。
1.3 调控Wnt/β-catenin信号通路
Wnt/β-连环蛋白(β-catenin)信号通路是调节骨代谢的经典信号通路,其在调节成骨细胞增殖、分化和骨形成过程中起着关键作用。Dickkopf 样蛋白1(Dickkopflike protein-1,DKK-1)和骨硬化蛋白(sclerostin,SOST)是Wnt 信号通路的抑制剂,可作为骨形成减少的标志物[19]。一项大鼠实验显示,灌胃艾司西酞普兰40 d后可提高股骨中DKK-1和SOST的水平,而卡比多巴可逆转艾司西酞普兰诱导的DKK-1和SOST水平升高[19]。由此可见,艾司西酞普兰通过抑制Wnt信号通路来减少骨形成,且该抑制作用可能是通过肠道5-HT 介导的。另有研究显示,氟西汀能抑制小鼠胚胎瘤细胞ATDC5 中Wnt/β-catenin 信号通路的激活,抑制蛋白聚糖的降解,上调软骨形成标志物性别决定区Y 框蛋白9 的表达,下调Wnt/β-catenin 信号转导标志物轴抑制蛋白2(axis inhibition protein 2,Axin2)和基质金属蛋白酶13 的表达;向骨关节炎模型大鼠关节腔内注射氟西汀可抑制其骨关节炎的进展[21]。由此可见,SSRI 可通过调控Wnt/β-catenin信号通路影响骨代谢。
1.4 调控BMP/Smad信号通路
研究显示,抗抑郁药发挥抗抑郁作用与其抑制海马体中骨形态发生蛋白(bone morphogenetic protein,BMP)的信号转导有关[22]。BMP 是介导成骨细胞增殖、分化和骨形成的重要调节信号,也是骨骼疾病的重要治疗靶点。Smad(Smad 1/5/8)依赖性途径是参与BMP 细胞内信号转导的主要途径。BMP可通过Smad信号通路调控重要成骨转录因子护骨因子(osteoprotegerin,OPG)的表达[23]。体外实验显示,氟伏沙明、舍曲林能够以剂量依赖的方式抑制MC3T3-E1细胞中的Smad 1/5/8信号通路,进而抑制BMP4刺激的OPG释放[23]。在小鼠抑郁症模型中,氟西汀可降低BMP4及其下游效应物磷酸化Smad 1/5/8(phosphorylated Smad 1/5/8,pSmad)的水平,提示其可抑制BMP 信号转导[22]。因此,SSRI 可能通过抑制BMP/Smad信号通路而减少骨形成。
SSRI影响骨形成的作用机制见图1。

图1 SSRI影响骨形成的作用机制
2 SSRI对骨吸收的影响及机制
相较于其他类型骨细胞,破骨细胞中SSRI 的作用靶点溶质载体家族6 成员4(solute carrier family 6 member 4,Slc6a4)的表达更强[24],提示SSRI对破骨细胞的影响可能是其影响骨稳态的重要机制。SSRI 可能通过下述不同途径影响破骨细胞的骨吸收功能。
2.1 影响破骨细胞分化和功能
舍曲林可使颅骨缺损模型小鼠的骨愈合减少,骨缺损处抗酒石酸酸性磷酸酶(tartrate-resistant acid phosphatase,TRAP)的阳性细胞数显著下降,提示破骨细胞活性降低[15]。该研究还表明,舍曲林可能直接抑制破骨细胞功能而不是通过细胞凋亡途径介导。另有研究表明,氟西汀、帕罗西汀、氟伏沙明可抑制小鼠破骨细胞分化和功能,而西酞普兰未显示出明显的抑制作用[25]。小鼠实验显示,用氟西汀20 mg/(kg·d)处理小鼠3 周后可观察到其通过抑制Ca2+/钙调蛋白-CREB-活化T 细胞核因子1(nuclear factor of activated T cells cytoplasmic 1,NFATc1)信号级联反应,以不依赖SERT 的方式直接抑制破骨细胞分化和功能[25]。然而,也有研究得出不同结论:以10 mg/(kg·d)氟西汀处理小鼠3 周后行骨折手术建立软骨内成骨模型和膜内成骨模型后进行愈伤组织TRAP 染色,结果显示氟西汀不干扰骨重塑过程中的破骨细胞活性[12]。如前所述,软骨内成骨和膜内成骨是骨重建过程中的两种骨折修复形式。在骨重塑过程中,骨吸收和骨形成紧密耦合;破骨细胞介导骨吸收,同时分泌耦合因子刺激成骨分化和骨形成[26]。该研究表明氟西汀对骨折愈合过程的负面影响可能主要是通过干扰成骨细胞而并非破骨细胞。不同的氟西汀药物浓度和实验模型可导致不同的实验结果。综上所述,SSRI可通过影响破骨细胞的分化与功能来影响骨吸收。
2.2 调控OPG/RANKL/RANK信号通路
RANKL 是激活破骨细胞生成的关键因子,OPG 可阻断RANKL 和核因子κB 受体活化因子(receptor activator for nuclear factor-κB,RANK)结合,从而抑制破骨细胞生成和骨吸收。有实验显示,艾司西酞普兰可增强大鼠血清RANKL活性,提高其股骨TRAP5b水平,提示骨吸收增强;联合卡比多巴不能改变艾司西酞普兰对骨吸收的影响,提示阻断肠源性5-HT不影响骨吸收[19]。另有研究显示,在大鼠类风湿性关节炎模型中,帕罗西汀可通过下调RANKL/OPG 信号通路介导抗风湿作用[27]。然而,Sheftel等[28]的研究显示,围产期小鼠接受舍曲林治疗后未观察到对RANKL/OPG信号通路的影响,骨吸收标志物Ⅰ型胶原交联羧基末端肽和相关基因亦未见改变。上述研究提示SSRI 可通过调控OPG/RANKL/RANK信号通路影响骨代谢,但不同药物的影响效果存在差异。
2.3 通过中枢5-HT信号介导促进骨吸收
Ortuño 等[25]的小鼠实验显示,短期使用氟西汀(3周)可产生抗骨吸收作用;而长期使用氟西汀(6 周)后,小鼠下丘脑中5-HT信号转导减少,交感神经输出增加,骨吸收增加抵消了氟西汀对破骨细胞的直接抑制作用,从而导致骨丢失;联用低剂量β 受体阻滞剂普萘洛尔(0.5 mg/d)可使交感神经输出正常化,预防氟西汀诱导的骨丢失。由此可见,根据用药时间长短,氟西汀对骨吸收呈现出双重作用,提示长期使用SSRI时,中枢5-HT可能在介导骨吸收方面起着重要的调控作用。
2.4 通过破坏骨髓脂肪组织中的鞘脂代谢促进破骨细胞生成
骨髓脂肪细胞(bone marrow adipocyte,BMA)在破骨细胞生成方面具有重要调节作用。Zhang等[29]的研究显示,长期服用氟西汀可抑制BMA 中鞘脂代谢途径的关键酶酸性鞘磷脂酶(acid sphingomyelinase,ASM),导致鞘磷脂(sphingomyelin,SM)分解为神经酰胺的过程受到干扰,引起代谢物1-磷酸鞘氨醇(sphingosine-1-phosphate,S1P)的水平降低,而S1P 的减少可介导环氧合酶2(cyclooxygenase-2,COX-2)/前列腺素E2(prostaglandin E2,PGE2)信号通路上调并诱导RANKL 的分泌增加,从而促进破骨细胞生成。该研究还显示,口服250 mg/(kg·d)L-丝氨酸(鞘脂合成前体)可预防重度抑郁症的绝经后妇女由于长期服用氟西汀(12 个月)导致的骨质流失。这提示抑制鞘脂代谢途径可能是SSRI影响骨吸收的潜在机制。
SSRI影响骨吸收的作用机制见图2。
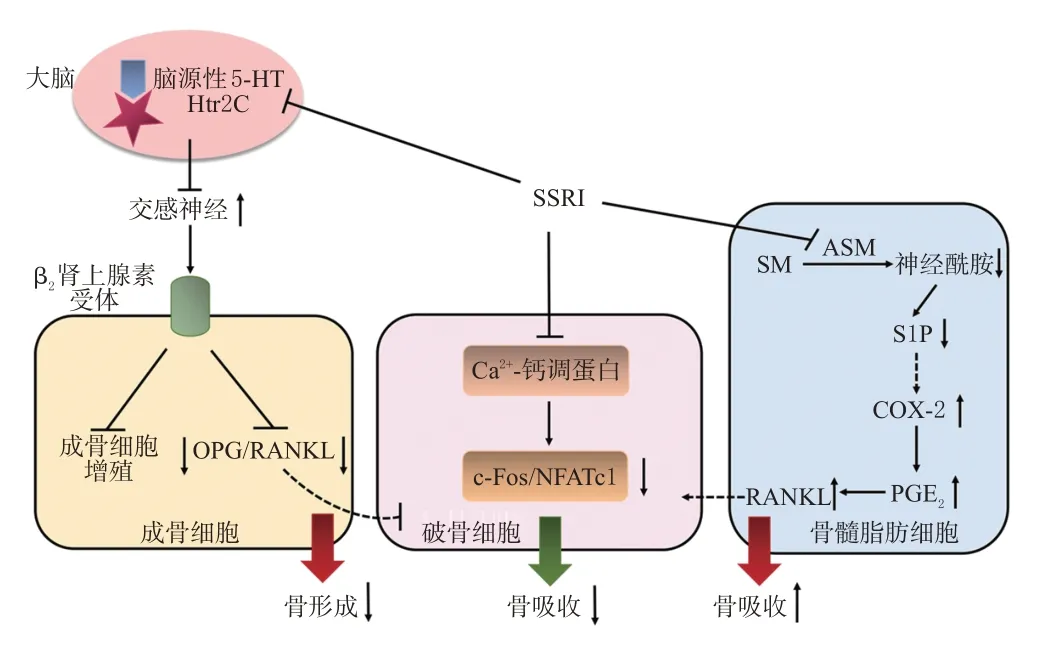
图2 SSRI影响骨吸收的作用机制
3 SSRI对间充质干细胞分化的影响
间充质干细胞具有多向分化潜能,能分化成多种细胞类型如脂肪细胞、成骨细胞、软骨细胞等,在骨代谢过程中起着重要作用。研究表明SSRI能够对间充质干细胞分化产生影响。
3.1 抑制间充质干细胞分化成骨
诱导细胞凋亡可能是SSRI影响间充质干细胞分化成骨的潜在机制。氟西汀可呈剂量依赖方式诱导大鼠脂肪间充质干细胞分化的骨祖细胞凋亡,且该作用与细胞上清液中的5-HT 浓度无关[30]。氟西汀对人脂肪干细胞增殖的影响还与时间相关,该药(10、20 µmol/L)处理细胞24 h后能促进细胞增殖,而处理48 h后则可抑制细胞增殖[31]。在该研究中,氟西汀还降低了成骨标志物RUNX2、ALP的表达水平并减少了矿化结节形成,提示氟西汀可抑制人脂肪干细胞向成骨分化。另据报道,人脂肪干细胞在接受氟西汀(5 µmol/L)治疗21 d 时,成骨分化早期标志物BMP2、RUNX2 以及晚期标志物Osterix、骨钙素、骨桥蛋白的表达增加,RANKL 的表达减少[32]。该研究与文献[31]得出不同结果可能与后者使用了较低剂量的氟西汀有关。此外,该结果与Ortuño等[25]的研究结果类似,提示氟西汀的短期使用可能对骨代谢有积极影响。
3.2 影响间充质干细胞向脂肪分化
实验证据表明,骨髓间充质干细胞向成骨分化和向脂肪分化存在反向关系,若诱导间充质干细胞向成骨分化,脂肪分化则会受抑制;但这一反向关系也存在例外情况,如BMP 信号转导可表现出促成骨分化和促脂肪分化的双重作用[33]。研究显示SSRI 可抑制间充质干细胞向成骨分化[30―32],意味着其可能诱导脂肪分化增加。但体外实验显示,氟西汀(10 μmol/L)可抑制人骨髓间充质干细胞向脂肪细胞的分化[34]。另一项研究结果类似:在人脂肪干细胞成脂分化过程中,氟西汀(10、20 μmol/L)可降低脂肪生成转录因子过氧化物酶体增殖物激活受体γ和脂肪酸结合蛋白的表达水平,并以剂量依赖的方式减少脂滴数量,提示氟西汀可抑制人脂肪干细胞向脂肪分化[31]。有研究显示,氟西汀可调控BMPSmad 1/5/8 信号通路[22―23],而该信号通路与间充质干细胞分化成骨和脂肪的过程密切相关[33],提示氟西汀对成骨分化和脂肪分化均可产生抑制作用的原因可能与其对该信号通路的抑制作用有关。但Ferroni 等[32]的研究显示,氟西汀(5 μmol/L)对人脂肪干细胞的成脂分化无显著影响,提示氟西汀对成脂分化的影响可能与其药物浓度有关。综上所述,SSRI可影响间充质干细胞向脂肪分化,同时影响骨代谢。
4 SSRI可调节炎症细胞因子的表达
SSRI具有一定的抗炎作用,该作用会对骨代谢产生影响。如帕罗西汀能减轻完全弗氏佐剂诱导的大鼠风湿性关节炎,其作用与降低血清炎症标志物白细胞介素6(interleukin-6,IL-6)、肿瘤坏死因子α、单核细胞趋化蛋白1以及氧化应激标志物丙二醛、还原型谷胱甘肽的水平有关[27]。在骨愈合过程中,早期炎症是骨愈合和骨整合的关键。研究显示,舍曲林可增加抗炎细胞因子IL-4、IL-10,同时减少促炎细胞因子IL-1β,导致骨愈合障碍[35]。由此可见,SSRI可通过调节炎症细胞因子的表达来影响骨代谢。
5 总结与展望
结合目前研究可知,短期使用SSRI 可能会对骨骼有积极影响,但长期使用SSRI 可能会导致骨骼健康问题。因此,对于长期使用SSRI的患者群体,应加强对骨骼健康的监测。不同SSRI类药物对骨代谢的影响存在差异,其中氟西汀对骨代谢的负面影响较大,其他SSRI类药物如艾司西酞普兰的影响可能较小。因此,替代药物治疗可能减少SSRI对骨骼的影响,对于骨折、骨不连或骨折高风险患者来说是更好的选择。
相关研究总体表明,SSRI 对骨代谢的影响错综复杂,其可通过骨形成、骨吸收、间充质干细胞分化、调节炎症细胞因子表达等多个方面对骨代谢产生影响。除了影响cAMP/PKA/CREB、Wnt/β-catenin、BMP/Smad、OPG/RANKL/RANK 等调控骨代谢的经典信号通路以外,SSRI 对骨代谢的影响还涉及中枢介导的作用。此外,除了上述信号通路外,参与调控骨代谢的其他信号通路还包括磷脂酰肌醇3激酶/蛋白激酶B信号通路、丝裂原活化蛋白激酶信号通路等,但就目前研究现状来说,SSRI 是否通过其他信号通路影响骨代谢则少见研究,有待后续进一步探讨。
值得注意的是,许多学者对SSRI 影响骨代谢机制的探究主要聚焦于其对成骨细胞的影响,但本研究发现SSRI 的作用靶点Slc6a4 在破骨细胞中的表达较在成骨细胞中更强,提示SSRI 影响破骨细胞的机制同样具有重要的研究价值。不同SSRI类药物对骨代谢的影响存在差异,但目前大部分学者研究SSRI 对骨代谢的影响主要针对氟西汀,而对其他SSRI 类药物相关机制的研究相对较少,有待进一步深入探究。此外,目前防治SSRI对骨代谢影响方面的相关药物研究较少,需要积极探索安全、有效的药物以防治SSRI 所带来的骨骼健康问题。
猜你喜欢
口腔医学(2021年10期)2021-12-02
中国骨质疏松杂志(2021年9期)2021-10-08
中国临床医学(2019年3期)2019-01-04
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
中华老年口腔医学杂志(2016年2期)2017-01-15
中国骨质疏松杂志(2016年1期)2016-01-29
中国病理生理杂志(2015年8期)2015-12-21
吉林大学学报(医学版)(2015年1期)2015-12-17
天津护理(2015年4期)2015-11-10
湖北科技学院学报(医学版)(2014年2期)2014-02-28
