
调节破骨细胞功能的相关信号分子的研究进展
2021-10-08 10:07赵常红李世昌李沛鸿翟晶
中国骨质疏松杂志 2021年9期
赵常红 李世昌 李沛鸿 翟晶
1.西北师范大学体育学院,甘肃 兰州 730030 2.华东师范大学体育与健康学院,上海 200241 3.甘肃省人民医院,甘肃 兰州 730000
破骨细胞是通过分泌组织蛋白酶K(cathepsin K, CtsK)、H+、Cl-和基质金属蛋白酶(matrix metalloproteinases, MMPs)来发挥蚀骨作用的一种大型多核细胞。破骨细胞前体细胞在M-CSF刺激下与相应受体c-Fms结合,并诱导RANK的表达,促进破骨细胞的生成[1]。通过RANKL刺激相应受体驱动下游信号因子,导致破骨细胞生成。本文通过近年来调控破骨细胞的分化和活性信号通路进行研究,探究其复杂关系,为防治骨质疏松症的药物研发提供可靠的分子生物学机制依据[2]。
1 RANKL/RANK/OPG信号通路
作为调控破骨细胞的主要信号通路,RANKL/RANK/OPG最为重要[3]。RANKL是TNF超家族的一种多肽II型跨膜蛋白,由成骨细胞产生,水解后以可溶性形式存在于细胞表面,能激活破骨细胞生成和迁移,增加骨吸收能力,然后周边的成骨细胞立即聚集填充骨陷窝形成新的骨表面,这就形成骨的重塑[4]。
OPG是一种可溶性蛋白,也由成骨细胞产生是RANKL的诱饵受体,能竞争地阻止RANKL与受体RANK的结合,能阻止破骨细胞的形成与蚀骨能力的增加[5]。OPG也属于TNFR超家族,也包含4个CRD,并且在CRD2和CRD3之间有一个铰链区域,可以结合到RANKL。因此,OPG通过竞争性地阻止RANKL和RANK的结合,调控破骨细胞的产生与活性,进而调节骨的构建与重塑,与RANK竞争性调节破骨细胞分化与活性,在骨重塑中具有十分重要的作用[6]。
RANK是RNAKL的信号受体,属于肿瘤坏死因子受体(TNFR)超家族分子的一种I型跨膜蛋白,由4个富含半胱氨酸的结构域(CRD)组成。RANK信号的激活可促进一些特异基因的表达,有利于促进破骨细胞分化、成熟,增加破骨细胞存活时间,激活破骨细胞骨吸收能力。RANK信号也能激活ERK-1激酶 ,ERK信号通路可促进破骨细胞形成[7]。RANK诱导的下游信号变化是判别破骨细胞活化的重要标识基因,尤其是TRAF6,RANKL与 RANK结合后,通过TRAF6可以将信号释放放大到下游 NFκB、MAPK、Src-AKT、Src-Ca2+信号通路中去,促进破骨细胞分化、成熟和蚀骨能力增加。
2 NF-κB信号通路
NF-κB是一个转录因子家族,包括RelA(也称为p65)、p50、p52、RelB和c-Rel,对破骨细胞的形成是必不可少的[8]。NF-κB p50和p52蛋白的共同表达,破骨细胞才能成熟发挥蚀骨功能。在破骨细胞分化过程中RANKL-RANK信号传递到下游通路主要通过IKKβ及其经典的NF-κB信号通路[9]。在RANKL刺激后,NF-κB、AP-1、NFATc2、ATF4和Jdp2被招募到NFATc1基因的启动子区,共同诱导NFATc1,这种转录因子的表达在RANKL被自身扩增机制刺激后大大上调,增强破骨细胞的生成。NF-κB p50和p52亚基的缺失导致破骨细胞的缺乏导致骨质疏松,随后观察到NF-κB对于RANKL和其他破骨细胞因子诱导的RANK表达的破骨细胞前体分化为TRAP+破骨细胞至关重要[10]。c-Fos和活化T细胞的核因子、NFATc1以及抑制NFATc1信号的抑制因子,也通过NF-κB信号蛋白、TRAF3和p100以及c-Fos/NFATc1信号转导抑制因子IRF8激活NF-κB信号转导,使破骨细胞皱褶的边缘膜下形成盐酸,溶解骨的矿物质成分,组织蛋白酶K被分泌以降解基质[11]。
3 MAPK /ERK信号通路
MAPK属于苏氨酸/丝氨酸蛋白激酶家族,由胞外信号调节激酶(Erk1/2)、p38-MAPKs(α/β/γ/δ)、c-Jun N末端激酶(JNK1、2、3)等组成。p38-MAPKs的激活在 RANKL诱导破骨细胞前体细胞向破骨细胞分化的过程中起到了重要作用。p38α在破骨细胞中表达最为丰富,这种信号分子的敲除导致破骨细胞的发生受损。单核细胞和巨噬细胞MAPK14f/fLysM-Cre小鼠中,p38被特异性地删除的小鼠显示出破骨细胞骨吸收减少,骨量增加[12]。很多研究表明,除了MAPK-JNK-AP-1信号外,还有ERK5、ERK7和ERK8也参与了RANKL对破骨细胞的分化调节,MAPK和Akt级联激活受M-CSF与其受体影响,从而使破骨细胞存活[13]。
ERK激活是成熟破骨细胞存活的核心,最近的研究表明p38 mapk-CREB通路在RANKL介导的破骨细胞分化中起重要作用。p38 MAPK抑制剂抑制TNF-α或RANKL诱导的CREB磷酸化,TNF-α或RANKL通过CREB磷酸化调节c-Fos和NFAT-c的表达,诱导破骨细胞分化[14]。最近的一项研究表明,Ambn通过抑制p38 MAPK-CREB磷酸化和下调c-Fos-NFAT c1轴来抑制RANKL诱导的破骨细胞分化[15]。p38 mapk在MAPK激酶激酶6(MKK6)激活后被激活,活化C激酶1受体(RACK1),在RANKL的反应下促进MKK6-p38 MAPK信号传导,并参与破骨细胞分化,ERK1/2和p38 mapk-CREB通路在骨稳态中起重要作用[16]。
4 M-CSF 信号通路
众所周知,单核/巨噬细胞系通过巨噬细胞集落刺激因子(M-CSF)的作用,是通过其受体c-fms诱导破骨细胞分化。GM-CSF和M-CSF是维持巨噬细胞数量和功能的重要因子。IL-34由成骨细胞产生,识别M-CSF受体(c-fms),从而促进破骨细胞的分化,如图1[17]。M-CSF通过与其受体 c-fms结合,诱导受体胞质端的七个酪氨酸残基进行磷酸化,在M-CSF诱导吞噬细胞和破骨细胞运动中,PI3K发挥非常重要作用。PI3K至少有3种不同的细胞表面受体下游效应子,包括 M-CSF受体、αvβ3以及 RANK。M-CSF能激活PI3K影响破骨细胞存活,也能调节破骨细胞肌动蛋白重塑,导致抑制膜皱褶、肌动蛋白环以及骨坑的形成。抑制PI3K的表达,既能有效抑制 RANKL及 M-CSF诱导的破骨细胞分化,也能降低其蚀骨能力。在 M-CSF/αvβ3整合素以及 DAP12-Syk信号通路下游,破骨细胞中调节细胞骨架重排的关键效应蛋白是 Vav3。Vav3-/-破骨细胞虽能正常分化,但不能形成胞足或密封圈,缺乏蚀骨能力[18]。
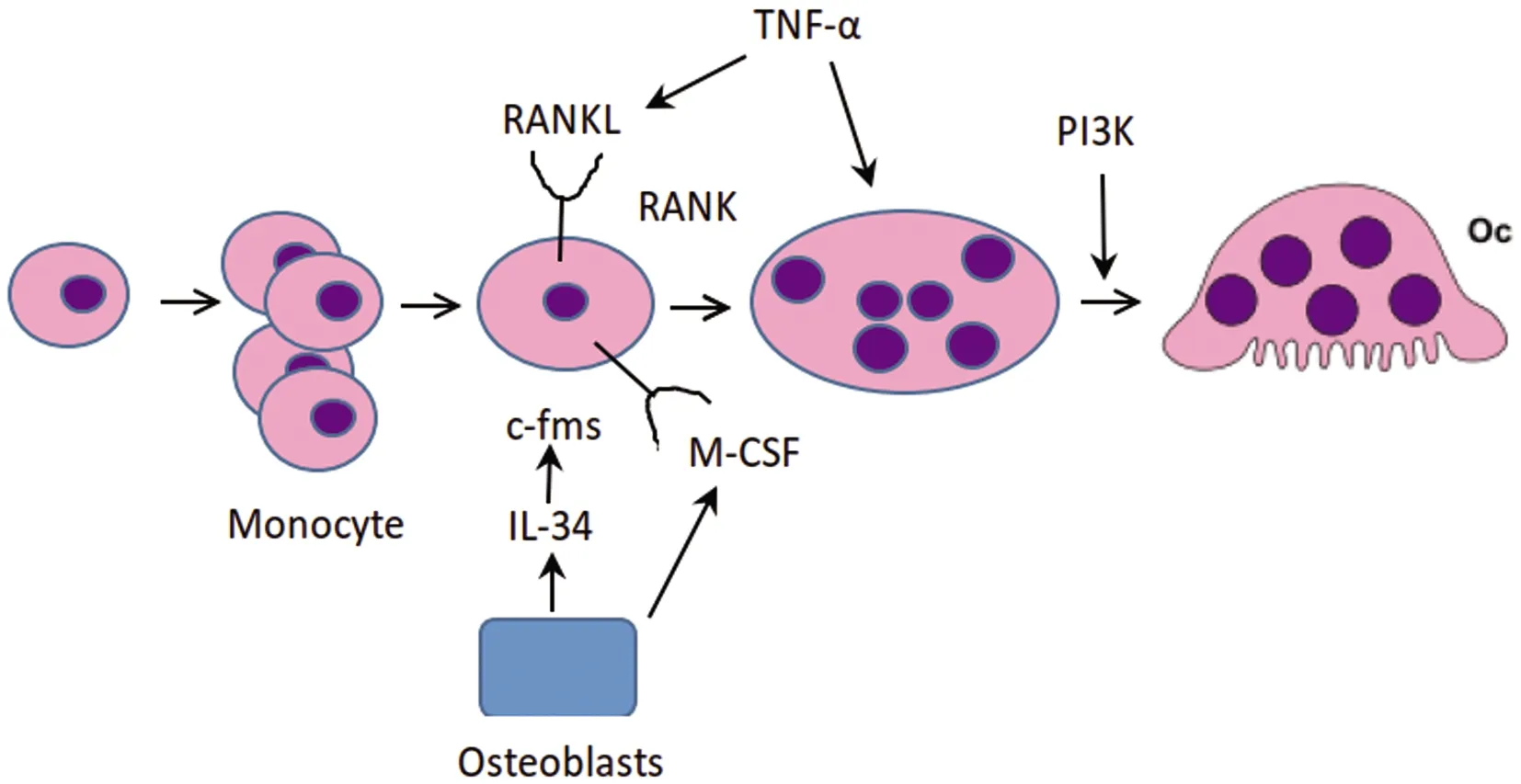
图1 M-CSF调控破骨细胞生成示意图Fig.1 Schematic diagram of osteoclast formation regulated by M-CSF
5 Ca2+信号通路
破骨细胞中的Ca2+信号是细胞分化、骨吸收和基因转录等多种细胞功能所必需的。研究发现,跨膜蛋白64(Tmem 64)是破骨细胞发生过程中Ca2+振荡的调节因子,Tmem 64缺乏可显著降低RANKL诱导的Ca2+振荡,导致Ca2+/钙调蛋白依赖性蛋白激酶(CaMK)IV和线粒体ROS减少,这两个都有助于实现破骨细胞形成所需的CREB活性[19]。Aβ增强NF-κB活性和IκB-α降解,激活ERK磷酸化,刺激钙振荡,从而导致破骨细胞活化过程中NFATc1表达上调,且激活转录因子ATF3参与钙信号传导激活破骨细胞的分化和活性[20]。调控Ca2+摄入主要由间质相互作用分子(stromal interaction molecule,STIM)及 Orai1Ca2+释放激活型钙调节子完成。STIM-1、2是一种多域、单通的跨膜蛋白,参与感知细胞内Ca2+水平的变化,并将细胞信号转导给Orai1通道蛋白,允许Ca2+内流。OCs中几种Ca2+通道的表达,发现STIM1在OCs早期高表达,TRPV4在OCs晚期高表达,且STIM1和TRPV4分别作为OCs分化早期和晚期钙通道可以作为破骨细胞发生或骨吸收的标志物[21]。研究发现,骨保护素通过Ca2+信号抑制破骨细胞分化和骨吸收,P2X7R-Ca2+-NFATc1信号通路在骨保护素诱导破骨细胞粘附结构损伤中起关键作用[22]。超过 24 h以上的RANKL刺激能使破骨细胞前体细胞发生Ca2+振荡,长时间低水平的Ca2+信号能激活NFAT,促进破骨细胞形成[23]。
6 Src、Akt信号通路
编码非受体酪氨酸激酶c-Src基因的缺失使成熟破骨细胞活动性降低,皱褶边缘及骨吸收的相关细胞骨架异常,不能有效发挥蚀骨功能。Src诱导Cox促进破骨细胞线粒体内提供高水平ATP,调节破骨细胞骨吸收活性。巨核细胞相关酪氨酸激酶(Matk),一种有效的c-Src抑制剂,对破骨细胞有抑制作用[24]。研究发现,RACK1-c-Src轴作为破骨细胞功能的关键调节器的作用[25]。Pkn3以Wnt5a-Ror2信号依赖方式与c-Src和Pyk2结合,从而增强破骨细胞c-Src的激酶活性,促进破骨细胞的骨吸收活性[26]。c-Src-/-小鼠破骨细胞外围胞足蚀骨带缺失,表现出粘附、延展以及迁移上的缺陷,不具有蚀骨能力[18]。Src蛋白一般与 TRAF6结合,诱导破骨细胞存活、骨架重排和迁移[8]。
研究证明,Akt信号调节破骨细胞的融合,Akt抑制可通过降低RhoA活性和增强Akt/GSK3β/NFATc1信号传导,触发破骨细胞数量和活性的急剧增加[27]。已有研究证明,壳寡糖能通过激活AKT促进破骨细胞的分化。T63预处理显著降低了Akt活性,抑制破骨细胞骨吸收,水苏碱(STA)抑制Akt信号传导,从而抑制活化NF-κB和NFATc1的活性,可减少破骨细胞数量并减轻骨量丢失[28]。苦皮藤醇(PIC)是一种天然的有机多酚二苯乙烯化合物,广泛存在于多种植物中,可通过抑制MAPK、NF-kB和AKT信号通路抑制RANKL诱导的破骨细胞发生和骨吸收,并促进成熟破骨细胞凋亡[29]。CAL-10对RANKL诱导的Akt-c-Fos/NFATc1信号级联抑制破骨细胞分化、迁移[30]。
7 PKC信号通路
PKC通路是破骨细胞重要的抑制性第二信使。研究发现,蛋白激酶 C(protein kinase, PKC)能调控破骨细胞分化与功能,如非典型 PKC(aPKC)支架蛋白 p62的突变会引起 Paget骨病,该病是一种破骨细胞异常激活导致的细胞功能障碍遗传性疾病,且RANK信号诱导形成aPKC、TRAF6及p62复合物,有利于破骨细胞形成[13]。研究已经发现,PKCβ通过参与M-CSF和RANKL的ERK信号通路,参与破骨细胞的形成和功能的调节,揭示了细胞外酸化通过PKC依赖性途径提高破骨细胞存活率,增加骨量丢失[31]。研究还发现p62相关的PKCζ在OCs过度活跃状态和NF-κB活化中的重要作用[32]。PKC-d的药物抑制和基因消融会损害体外破骨细胞骨吸收,破骨细胞前体的迁移依赖于通过整合素avβ3绕过RhoA和Rac1介导的PI3K/PKCa-PKCd信号传导,而成熟破骨细胞的迁移依赖于整合素avβ3介导的PI3K/PKCaPKCd/RhoA-Rac1信号轴[33]。PKC-δ也是骨代谢的重要调节因子,在破骨细胞中PKC-δ的消融导致雄性小鼠骨小梁和皮质骨体积增加,而雌性小鼠则没有观察到骨量表型。伴随着破骨细胞数量和表面积的减少,组织体内PKC蛋白水平以及破骨细胞的形成和吸收减少,是雄性特有的方式[34]。
8 Ig-like 受体信号通路
Ig-like的两个受体接头蛋白DAP12和FcRγ,表达在NK和骨髓细胞膜上,并通过ITAM结构域传递信号,这条信号通路是RANK的共调信号,Ig-like受体在破骨细胞分化中起重要作用。研究发现,细胞表面ITIM含有Ig样受体调控破骨细胞形成,抑制性Ig样受体招募Src同源2结构域的酪氨酸磷酸酶1(SHP-1)在破骨细胞前体细胞上表达,并在破骨细胞的发育中起调节作用,调节这些调节受体可能是控制各种骨骼系统疾病和炎症性关节炎[35]。人破骨细胞相关受体(OSCAR)是一种免疫球蛋白(Ig)样胶原受体,在破骨细胞发生过程中对破骨细胞上调,并在多种髓样细胞中表达。此外,CLP肽作为破骨细胞生成的抑制剂,发现需要40个氨基酸的肽段才能在体外充分抑制破骨细胞生成。这些发现为OSCAR的胶原识别模式以及合成肽基质因子在破骨细胞生成抑制中的应用提供了有价值的结构见解[36]。
9 结论
骨组织代谢的吸收和形成之间处于微妙动态平衡。骨吸收依赖于破骨细胞相关信号的完美时空协调。破骨细胞在骨量丢失疾病如骨质疏松症中发挥重要作用,了解和研究这些信号分子对破骨细胞的调节作用,有助于调控破骨细胞的分化和活性,利用这些转运机制有助于开发新的防治骨质疏松症等骨代谢疾病的抗骨吸收新疗法,以破骨细胞为靶点的药物,在防治骨量丢失相关疾病方面具有重要意义。
猜你喜欢
清华金融评论(2022年4期)2022-04-13
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
国际放射医学核医学杂志(2021年10期)2021-02-28
山东医药(2020年36期)2020-12-31
房地产导刊(2020年7期)2020-08-24
中国临床医学(2019年3期)2019-01-04
中国骨质疏松杂志(2016年1期)2016-01-29
中国医药生物技术(2015年4期)2015-12-26
中国药理学通报(2014年2期)2014-05-09
