
超高效液相色谱-串联质谱法测定化妆品中52种抗感染类药物
2023-11-29 01:42张丽蓉肖紫芬谢宋阳毛林芳
分析测试学报 2023年11期
张丽蓉,肖紫芬,谢宋阳,毛林芳,王 慧,陈 硕*
(1.福建省食品药品质量检验研究院,福建 福州 350001;2.中国食品药品检定研究院,北京 100050)
喹诺酮类、林可酰胺类、硝基咪唑类等抗感染类药物,大多属于临床使用的处方药,可用于治疗皮肤病(除螨、祛痘等)。而长期使用这些药物可能刺激皮肤,引起接触性皮炎,甚至对人体脏器造成一定伤害[1-2]。我国化妆品监管现行标准《化妆品安全技术规范》2015年版(简称《规范》)[3]及欧盟化妆品法规均明确将此类药物列为化妆品禁用原料。受经济利益的驱使,部分生产企业非法添加《规范》检验方法监测范围外的其他抗感染类药物,以规避化妆品技术规范及法律法规的监管。为保障消费者安全用妆,建立涵盖药物种类更齐全、操作更便捷高效的检测方法尤为重要。
现有文献报道化妆品中抗感染类药物的检测方法有高效液相色谱法[4-5]、高效液相色谱-质谱法[2,6-11]、超高效液相色谱-四极杆-飞行时间高分辨质谱法[12-13]、化学发光酶联免疫法[14]和气相色谱-质谱法[15]。目前《规范》中抗感染类药物的检测方法有3种,包括氟康唑等9种组分检测方法、依诺沙星等10种组分检测方法以及化妆品中抗感染类药物检测方法,共涵盖40种抗感染类药物。检测方法的相对分散性致使检验工作重复、方法耗时、成本高,故开发一种同时测定化妆品中更多种抗感染类药物的方法具有重要意义。本文考虑将《规范》中上述3 个方法合并,同时将临床中常用的抗感染类药物,如莫匹罗星、红霉素、喹诺酮类(加替沙星、那氟沙星、司帕沙星、洛美沙星)、磺胺类(磺胺甲嘧啶、磺胺噻唑、磺胺苯酰、磺胺二甲嘧啶)、硝基呋喃类(奥硝唑、替硝唑)抗菌药物以及近两年国家药监局发布的不合格化妆品通告[16]中祛痘类产品检出的西咪替丁并入筛查,以期加大监测范围。此外,《规范》中抗感染类药物检测方法存在以下问题:①磺胺类药物经0.5%甲酸乙腈溶液提取后,稳定时间较短,可能导致定量结果不准确;②克林霉素磷酸酯经饱和氯化钠溶液分散后提取不完全,可能造成添加量不高的样品出现假阴性结果。
本文建立了测定化妆品中52 种抗感染类药物的超高效液相色谱-串联质谱(UPLC-MS/MS)方法,针对《规范》存在的上述两个问题进行优化改进,同时增加莫匹罗星等13种有违禁添加风险的抗感染类药物,扩大了检测范围,可为化妆品中抗感染类药物违禁添加的质量安全监管提供技术支持。
1 实验部分
1.1 仪器与试剂
UPLC-I-Class Plus 超高效液相色谱仪、Xevo TQ-S 三重四极杆质谱仪(美国沃特世公司);电子分析天平(瑞士梅特勒-托利多公司);超声波清洗仪(美国IKA 公司);高速冷冻离心机(日立Hitach 公司);Arium Combort超纯水仪(德国赛多利斯公司)。
磺胺吡啶、莫匹罗星、甲硝唑、磺胺甲二唑、盐酸萘替芬、磺胺甲氧嗪、西咪替丁、螺内酯、咪康唑、磺胺氯哒嗪、克拉霉素、磺胺嘧啶、酮康唑、罗红霉素、磺胺二甲嘧啶、替硝唑、氯霉素、磺胺噻唑、磺胺苯酰均购于中国食品药品检定研究院,纯度均大于96.0%;氧氟沙星、氟罗沙星、环丙沙星、磺胺甲唑、依诺沙星、盐酸沙拉沙星、盐酸双氟沙星、盐酸四环素、恩诺沙星、米诺环素、呋喃它酮、氟康唑、奥硝唑、土霉素、盐酸金霉素、阿奇霉素、灰黄霉素均购于Dr.Ehrenstorfer 公司,纯度均大于95.0%;磺胺甲嘧啶、联苯苄唑、益康唑、红霉素均购于Bepure 公司,纯度均大于98.0%;盐酸多西环素、克霉唑、林可霉素、克林霉素、克林霉素磷酸酯、培氟沙星、诺氟沙星、加替沙星、那氟沙星、司帕沙星、洛美沙星均购于CATO 公司,纯度均大于97.0%;莫西沙星购于EP 公司,纯度为100.0%;乙腈、甲醇(色谱纯,德国默克公司);甲酸(色谱纯,阿拉丁);实验用水为超纯水。
1.2 实验方法
1.2.1 标准溶液的制备精密称取52种抗感染类药物标准品各10 mg,分别置于10 mL量瓶中,用甲醇溶解并定容(部分标准品用水先溶解),配制成质量浓度约为1 mg/mL 的标准储备溶液。分别精密移取各标准储备溶液适量,置于同一量瓶中,用甲醇稀释并定容,配制成质量浓度约为1 μg/mL 的混合标准溶液。
1.2.2 基质标准溶液的制备准确称取基质空白样品7份(0.2 g/份),置于15 mL离心管中,分别加入混合标准溶液适量,按供试品溶液制备步骤处理,配制成质量浓度为1~50 ng/mL的基质标准溶液。
1.2.3 供试品溶液的制备准确称取0.2 g 样品,置于15 mL 离心管中,加入乙腈5 mL,涡旋30 s,超声提取30 min,摇匀,以10 000 r/min 于0 ℃冷冻离心10 min,溶液转移至10 mL 量瓶中,加水稀释定容,摇匀,过滤,即得。
1.2.4 色谱条件色谱柱:Waters HSS T3(2.1 mm×100 mm,1.8 μm);流动相:A 相为0.1%甲酸乙腈,B 相为0.1%甲酸水;柱温:40 ℃;进样量:2 μL;流速:0.3 mL/min。梯度洗脱程序:0~3 min,2% ~15% A;3~9 min,15%~50% A;9~14 min,50%~99% A;14~17 min,99% A;17~17.5 min,99%~2% A;17.5~22 min,2% A。
1.2.5 质谱条件离子源:电喷雾电离源(ESI),检测方式:多反应监测(MRM);毛细管电压:3.0 kV;离子源温度:150 ℃;脱溶剂气温度:500 ℃,脱溶剂气流量:1 000 L/h;质谱参数见表1。
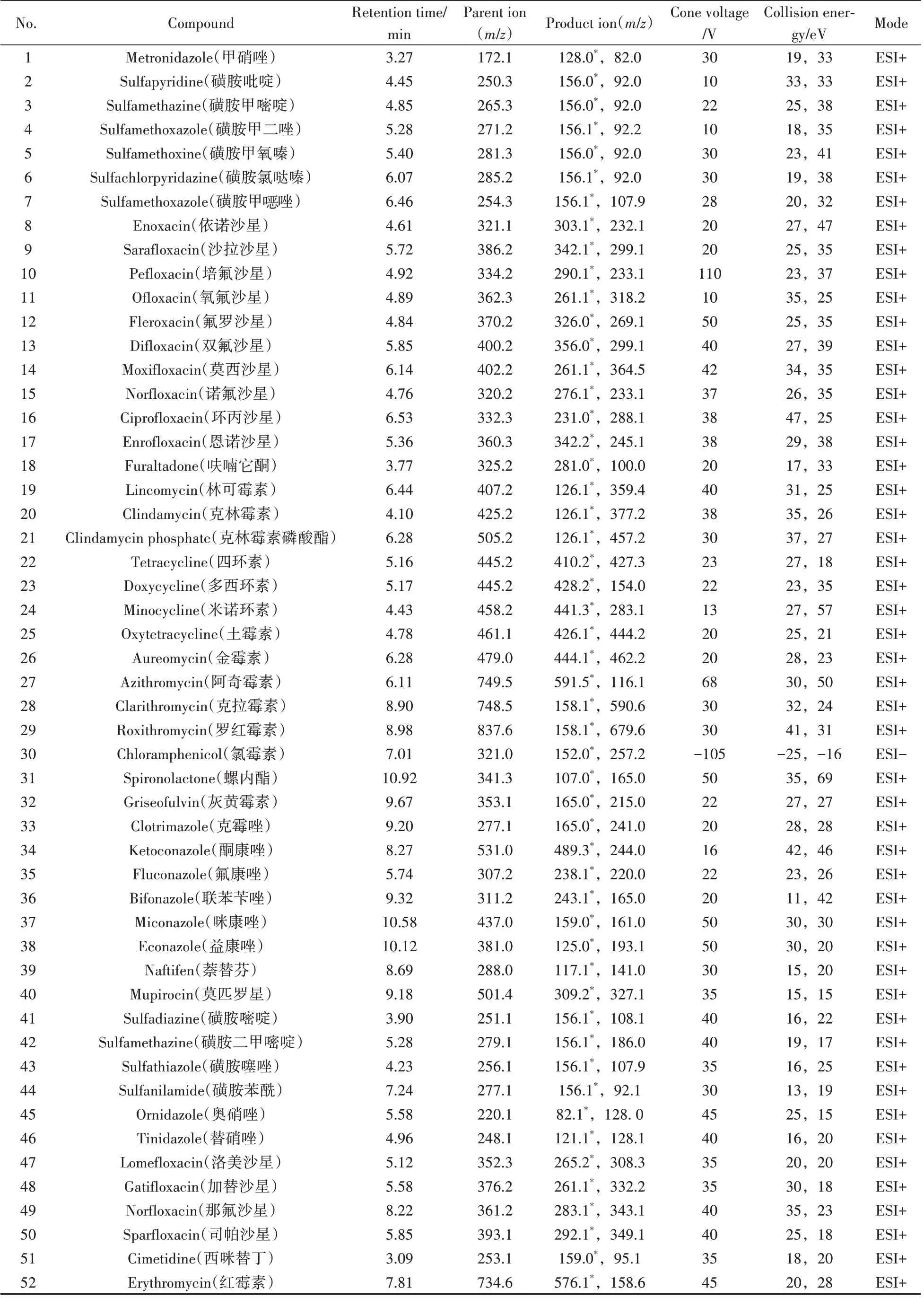
表1 52种抗感染类药物的质谱参数Table 1 MS parameters of 52 anti-infective drugs
2 结果与讨论
2.1 色谱柱的选择
本研究的抗感染类药物成分多、极性差异大,因此选择通用型反相色谱柱进行分离,以获得较好的色谱保留和峰形。对比了Waters HSS T3(2.1 mm×100 mm,1.8 μm)及Waters BEH C18(2.1 mm×100 mm,1.7 μm)2 款色谱柱的分离效果,发现两者响应基本一致,且均能实现对目标分析物的较好分离,但HSS T3 柱对极性较强化合物有更好的色谱保留,更适合分离极性差异大的化合物。进一步比较了Waters HSS T3(2.1 mm×100 mm,1.8 μm)、Agilent ZORBAX SB-Aq(2.1 mm×100 mm,1.8 μm)和CAPCELL PAK C18(2.0 mm×150 mm,2.7 μm) 3 款色谱柱的分离效果,结果显示HSS T3 柱与CAPCELL PAK C18柱的响应强于Agilent ZORBAX SB-Aq 柱,但HSS T3 柱和Agilent ZORBAX SB-Aq 柱的峰形较优。综合比较,最终选择Waters HSS T3(2.1 mm×100 mm,1.8 μm)进行分离测定。
2.2 流动相的选择
考察了常用的流动相体系甲醇水、乙腈水对待测组分的分离情况,结果显示乙腈水对待测组分的适用性高于甲醇水。因乙酸铵能屏蔽硅醇基,起到改善峰形的作用,且与质谱的兼容性较好,本研究拟采用乙酸铵改善个别成分的峰形。但实验发现其对化合物离子化具有抑制作用,尤其对四环素类成分的离子化抑制作用较明显,综合考虑后选择不加乙酸铵。在ESI+模式下,加入少量甲酸能促进化合物电离后加氢从而增强响应,因此考察了流动相中加入不同体积分数甲酸(0.05%、0.1%、0.2%、0.5%)时目标分析物的分离效果。结果显示,氯霉素的响应在加甲酸的流动相体系中基本不受影响,其余待测组分的响应随着流动相中甲酸体积分数的增大而增强,由于高浓度甲酸会损伤仪器,最终选用0.1%甲酸水-0.1%甲酸乙腈作为流动相。52 种抗感染类药物混合标准溶液(10 ng/mL)的总离子流图见图1A~E,阳性样品的典型离子流图见图1F。
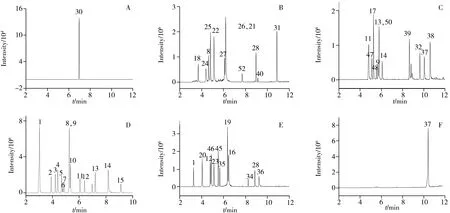
图1 52种抗感染类药物混合标准溶液(A~E)及阳性样品(F)的总离子流图Fig.1 Total ion current chromatograms of 52 anti-infective drugs(A-E) and a positive sample(F)the peak numbers denoted were the same as those in Table 1
2.3 前处理方法的优化
考察了分别以甲醇和乙腈作为提取溶剂时的效果,结果显示,甲醇作为提取溶剂时起泡及乳化现象较明显,而乙腈作为提取溶剂时不存在上述现象,且流动相为乙腈,故选择乙腈作为提取溶剂以降低溶剂效应。实验发现,采用《规范》中的前处理方法(样品经饱和氯化钠溶液涡旋后用0.5%甲酸乙腈溶液进行提取)存在以下问题:①供试品溶液中的磺胺类药物在酸性条件中不稳定,6 h 后响应值显著下降;②克林霉素磷酸酯受脂水分配系数的影响,只有约10%进入乙腈层,其余在饱和氯化钠层。针对上述问题,本研究不使用饱和氯化钠溶液分散样品,直接使用乙腈进行样品提取,磺胺类药物及克林霉素磷酸酯的回收率均能达到90%以上。
为降低基质效应,考察了Waters、艾杰尔等品牌的HLB固相萃取小柱(60 mg、200 mg规格)的净化效果,发现西咪替丁等强极性成分无法保留;采用岛津SHIMSEN Q 小柱进行净化处理时,克林霉素磷酸酯和四环素类成分损失较多,回收率低。实验最终采用乙腈对样品进行超声提取,并在0 ℃冷冻离心后取上清液加水稀释的前处理方法,既能沉淀蛋白和去除油脂类杂质,又不影响回收率。
2.4 基质效应
化妆品的基质复杂,采用乙腈进行提取后,基质共提取物竞争电离,从而影响待测组分的离子化程度,形成基质效应(ME),影响定量结果的准确性。ME按下式计算:ME(%)=(基质标准曲线斜率/溶剂标准曲线斜率-1)×100%[17],当ME为负值,说明存在基质抑制效应,正值为基质增强效应。当|ME|≤20%,表示存在较弱的基质效应;当20%<|ME|≤50%,表示存在中等的基质效应;当|ME|>50%,表示存在较强的基质效应。52种抗感染类药物的ME值见表2。结合前处理考察的情况,采用基质标准工作曲线进行定量分析以减弱基质效应的影响。
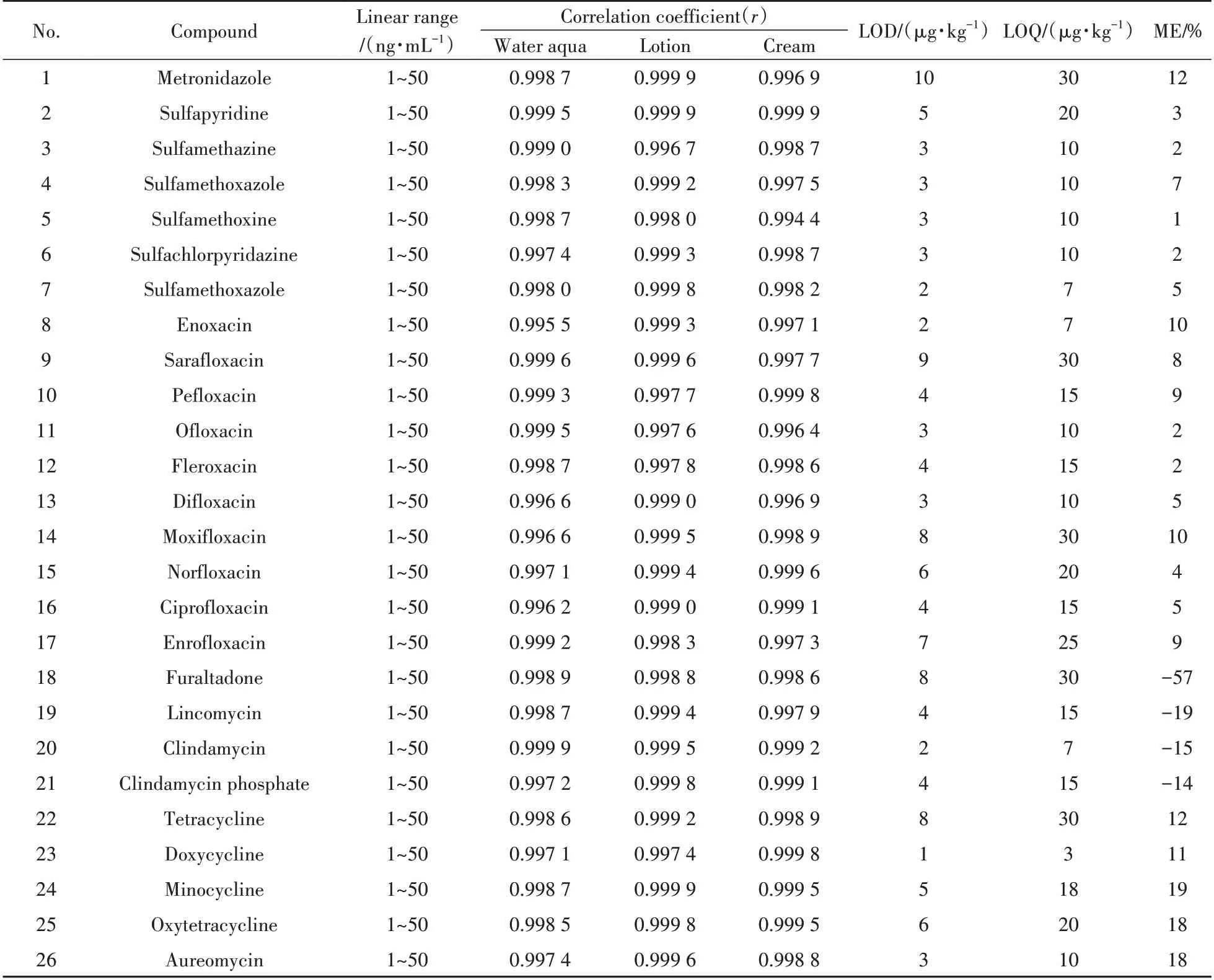
表2 52种抗感染类药物的线性关系、检出限、定量下限与基质效应Table 2 Linear relations,limits of detection,limits of quantitation and matrix effects of 52 anti-infective drugs
2.5 方法学验证
2.5.1 线性、检出限与定量下限按“1.2.2”方法配制3 个基质(水剂、乳液、膏霜)标准工作溶液,采用本方法进行测定,以各药物的质量浓度为横坐标,峰面积为纵坐标进行拟合。由表2 可知,52种抗感染类药物在1~50 ng/mL范围内呈良好的线性关系,相关系数(r)均大于0.99。取基质效应最大的膏霜类基质阴性样品,添加适量混合标准溶液,按供试品溶液制备同法操作,以3 倍信噪比计算检出限(LOD),10倍信噪比计算定量下限(LOQ),得到52种抗感染类药物的检出限和定量下限分别为1~10 μg/kg和3~35 μg/kg。
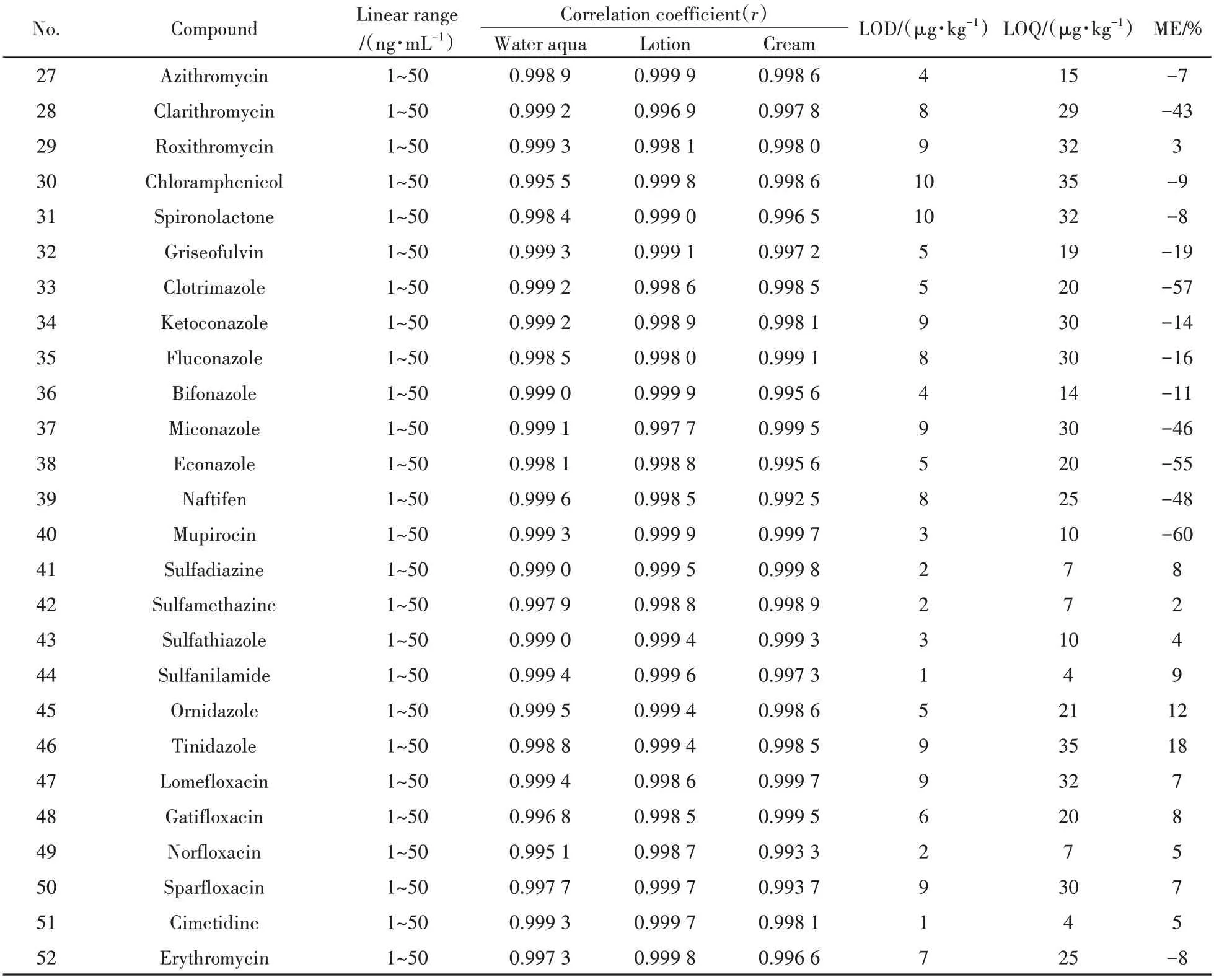
(续表2)
2.5.2 加标回收率与相对标准偏差取水剂、乳液、膏霜3 类基质阴性样品,按0.04、0.08、0.4 mg/kg 3个水平添加52种抗感染类药物的混合标准溶液,与供试品溶液制备同法操作,每个浓度平行实验3 次。52 种抗感染类药物在水剂中的平均回收率为86.5%~116%,相对标准偏差(RSD)为1.2%~7.6%;乳液中的平均回收率为81.5%~116%,RSD为0.94%~9.2%;膏霜中的平均回收率为81.2%~116%,RSD为1.2%~8.9%。
2.6 样品测定
应用本方法对市售的50批祛痘类化妆品及50批儿童护肤类产品进行测定,结果显示1批儿童护肤类产品检出咪康唑,含量为0.68 mg/kg,阳性样品的总离子流图见图1F。
3 结 论
本文将《规范》中3 个抗感染类药物测定的标准方法合并,并在此基础上增加莫匹罗星等13 种临床常用外用药,提高了工作效率,节约了资源成本,扩大了检测范围。相较于《规范》中抗感染类药物的现行标准,本方法在保证结果准确的前提下,通过对前处理过程进行优化,有效避免了磺胺类药物降解和克林霉素磷酸酯提取不完全的问题,同时检出限和定量下限均低于《规范》的现行标准,提高了方法灵敏度。本方法高效便捷,灵敏度高,分析范围广,具有良好的选择性,可为化妆品中抗感染类药物违禁添加的质量安全监管提供较好的技术支持。
猜你喜欢
武警医学(2018年10期)2018-11-06
中国蜂业(2018年4期)2018-05-09
中国化妆品(2017年12期)2017-06-27
中国化妆品(2017年12期)2017-06-27
健康女性(2017年3期)2017-04-27
当代化工研究(2016年6期)2016-03-20
湖南农业(2016年12期)2016-03-10
无机化学学报(2014年3期)2014-02-28
无机化学学报(2014年3期)2014-02-28
河南科技(2014年12期)2014-02-27
