
禾谷镰刀菌侵染条件下小麦酵母双杂交文库的构建及分析
2023-11-28 04:58刘海燕赵维萍
滁州学院学报 2023年5期
刘 洋,刘海燕,吴 旭,方 舟,王 君,赵维萍,蔡 华
小麦(TriticumaestivumL.)作为一种重要的粮食作物,能够为全球40%以上的人口提供淀粉、蛋白质和膳食纤维等营养物质[1]。随着全球人口的激增和人们生活水平的提高,对小麦产量和品质的需求也在不断增加。然而,小麦在生产过程中会遭遇来自不同生物和非生物胁迫因素的持续威胁,导致产量减少、品质降低。其中,在全球范围内,由禾谷镰刀菌(Fusariumgraminearum)引起的小麦赤霉病是危害最大的真菌病害之一,可造成高达50%的产量损失,并且病原菌产生的脱氧雪腐镰刀菌烯醇(DON)等真菌毒素还会污染小麦籽粒,导致品质下降[2]。因此,提高小麦抗赤霉病能力、降低赤霉病的感染面积和发病程度能够有效增加小麦产量和减少毒素积累。
在小麦抗性基因库中不断地挖掘抗赤霉病相关基因,探明F.graminearum侵染小麦的机制和小麦抗赤霉病的分子机制,是利用分子技术培育抗赤霉病小麦新品种的基础。随着小麦参考基因组测序的完成,蛋白质功能组学研究也已成为热点。由于生物体内各种生命活动基本上都是通过蛋白质与蛋白质之间的相互作用来完成的,因此找出与未知蛋白互作的蛋白,并构建互作网络,能够对探究未知蛋白质的功能提供线索。酵母双杂交技术是研究生物体内蛋白互作的最常见的方法之一,最早由Fields和Song在研究真核生物细胞转录因子(酵母转录因子GAL4)性质时提出了核体系酵母双杂交[3];后来,Johnsson和Varshavsky又根据酵母细胞中分裂泛素的结构与性质发明了分裂泛素系统膜体系酵母双杂交技术[4]。通过酵母双杂交技术筛选互作蛋白来揭示基因蛋白功能及解析其作用机理的前提条件是需要构建高质量的酵母双杂交文库[5-6]。因此,为进一步解析小麦防御赤霉病的机制,构建F.graminearum侵染下小麦的cDNA文库就显得十分重要。

A.总RNA电泳图;B.分离纯化后mRNA电泳图;C.合成dscDNA电泳图图1 禾谷镰刀菌侵染条件下小麦穗部总RNA、mRNA 与dscDNA琼脂糖电泳图
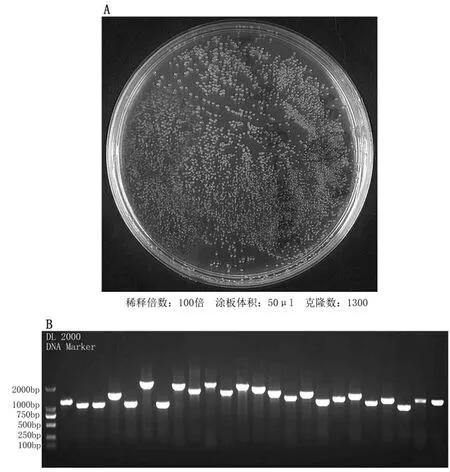
A.初级文库库容量鉴定;B.初级文库插入片段重组率鉴定图2 禾谷镰刀菌侵染条件下小麦穗部酵母双杂交初级文库构建及质量评价
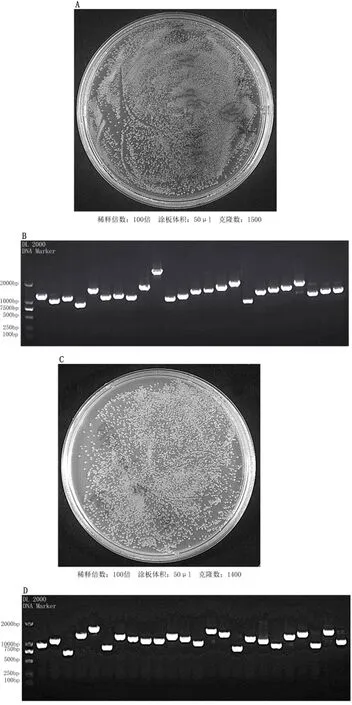
A.核体系次级文库库容量鉴定;B.核体系次级文库插入片段重组率鉴定;C.膜体系次级文库库容量鉴定;D.膜体系次级文库插入片段重组率鉴定图3 禾谷镰刀菌侵染条件下小麦穗部酵母双杂交次级文库构建及质量评价

A.文库滴度鉴定;B.酵母克隆鉴定图4 核体系次级文库质粒转化Y187酵母菌株
目前cDNA文库的构建主要采用SMART技术,将合成的cDNA和线性化处理的载体共同转化到酵母细胞,在体内重组酶的作用下让cDNA和载体进行重组连接形成完整的环状重组质粒,这项技术由于经过了PCR扩增,因此仅需要使用少量的cDNA就可合成全长克隆并获得完整的cDNA文库[7-8]。利用SMART技术徐新凤等构建了不结球白菜酵母杂交cDNA文库[9],王巧慧等构建了白粉菌侵染条件下小麦酵母双杂交cDNA文库[10],滕珂等构建了日本结缕草衰老叶片全长cDNA文库[11],徐晓兰等构建了赤芝酵母单杂交cDNA文库[12]。除SMART技术之外,Gateway技术也常用于构建cDNA文库,它是利用λ噬菌体位点特异重组系统的BP和LR双向反应,首先利用BP重组反应将cDNA克隆到入门载体上(如pDONR222)形成入门文库,然后再根据不同的目的选择不同的表达载体,通过LR重组反应让cDNA快速且定向地重组到任何Gateway化的载体上,最终形成各种所需的表达文库[13-14]。陈玉婷等采用Gateway技术构建了受青枯菌诱导的花生根酵母双杂交文库[15];李立群等以中国春小麦不同组织和不同时期籽粒为材料,利用Gateway技术构建了小麦酵母杂交cDNA文库[16];类似的,杨少丽和秦松以成熟期菊芋块茎为材料,通过Gateway技术将全长cDNA与pDONR222载体重组构建出菊芋非剪切型全长cDNA文库[17]。
本研究以接种F.graminearum后不同时间点的安农1589小麦穗部为材料,基于Gateway技术,采用CloneMinerTMII试剂盒构建小麦cDNA初级文库,然后通过LR重组构建出酵母双杂交核体系cDNA次级文库和膜体系cDNA次级文库,并将核体系cDNA文库质粒转化进Y187酵母菌株获得核系统酵母双杂交文库。本研究构建的文库以期为筛选小麦抗赤霉病蛋白的互作蛋白提供丰富资源,为研究小麦与禾谷镰刀菌互作分子机制提供有力支撑。
1 材料与方法
1.1 材料
1.1.1 试验材料
以中抗赤霉病小麦品种安农1589为试验材料,将小麦种子播种于花盆中,然后将花盆半埋于小麦育种试验田,正常田间管理。待小麦开花期,从试验田挖出花盆,移放到温室内,用移液枪吸取15μL的禾谷镰刀菌(混合菌株)分生孢子悬浮液注射入小麦穗部的每朵小花的内外稃之间,所有小花接种完后套上杂交袋,并喷水保湿,于接种后24h、36h、48h和72h进行取样,样品经液氮急速冷冻后保存于-80℃冰箱备用。
1.1.2 菌株和载体
酵母菌株Y2H Gold、酵母菌株NMY51、大肠杆菌DH10B、载体pDONR22、pGADT7-DEST和pPR3-N-DEST均购自Clontech公司;大肠杆菌感受态细胞DH5ɑ、酵母感受态细胞Y187均购自上海唯地生物技术有限公司。
1.1.3 主要试剂
Oligotex mRNA Midi Kit 购自天根生化科技(北京)有限公司;CloneMineTMII cDNA Library Construction Kit、Superscript®Double-Stranded cDNA Kit、Gateway®BP Clonase®II enzyme mix、GatewayTMLR ClonaseTMII enzyme mix均购自美国Invitrogen公司;TaKaRa MiniBEST Plant RNA Extraction Kit、CHROMA SPINTM+TE-1000 Columns离心柱购自宝生物工程(大连)有限公司;2×Rapid Taq Master Mix、FastPure Plasmid Mini Kit均购自南京诺唯赞生物科技股份有限公司;氯化钠(NaCl)、琼脂糖(Agarose)、酵母提取物、琼脂粉、胰蛋白胨等均购自生工生物工程(上海)股份有限公司;所有引物合成均由生工生物工程(上海)股份有限公司完成。
1.2 方法
1.2.1 小麦穗部总RNA的提取
取前期冻存的经禾谷镰刀菌接种处理后不同时期的小麦穗,采用TaKaRa MiniBEST Plant RNA Extraction Kit提取小麦穗部总RNA,用1.5%浓度的Agarose胶电泳检测提取的RNA的完整性,并用Nano-Drop2000微量核酸分析仪检测RNA的质量和浓度,然后取接菌处理不同时期的RNA进行等比例混合用于建库使用。
1.2.2 小麦穗部mRNA的分离与纯化
参考Oligotex mRNA Midi Kit说明书从小麦穗部总RNA中分离并纯化mRNA。分离、纯化后取10μL产物用1%浓度的Agarose凝胶进行电泳质检,根据电泳条带的弥散程度及高亮度所在区域来判断。
1.2.3 安农1589穗部cDNA初级文库构建及质量鉴定
参照CloneMinerTMII 说明书进行。
(1)双链cDNA(dscDNA)的合成
取纯化后的mRNA按照SuperScript®Double-Stranded cDNA Kit试剂盒进行反转录合成dscDNA。在无菌无酶的PCR管中依次加入5μg mRNA、7μL DEPC water、1μL biotin-attB2-Oligo(dT)Primer、1μL dNTPs,混合均匀后置于PCR仪中:70℃ 5min→45℃ 2min。向上步反应液中加入4μL 5×First Strand Buffer、2μL DTT、5μL SuperScript III RT酶,至总体积20μL ,混合均匀后置于PCR仪中:45℃ 20min→50℃ 20min→ 55℃ 20min。反应结束后获得cDNA第一链。
向上述含有cDNA第一链的反应液中按照表1依次加入各成分,混合均匀后置于PCR仪中在16℃条件下反应2h。然后,加入2μL T4 DNA Polymerase,在16℃条件连接10min。连接完成后,转移至1.5mL无菌无酶离心管中,并加入10μL 0.5M EDTA(pH8.0),补充dd-water 至300μL ,再向上述溶液中再加入300μL 酚:氯仿:异戊醇(25:24:1),轻轻上下颠倒混匀30s,冰上静置1min。静置后于高速离心机中14000rpm室温离心5min,转移上清液于新的1.5mL无菌无酶离心管中,依次加入1μL Glycogen、150μL 5M NH4OAc、1125μL 无水乙醇,混合后置于超低温冰箱(-80℃)静置15min,再置于离心机4℃、14000rpm条件下离心25min,除去上清,保留沉淀。用1mL 70%乙醇溶液洗涤沉淀两次:4℃、14000rpm、3min,去除上清液。沉淀在室温条件下晾干后,再加入104μL DEPC water溶解cDNA,即可获得dscDNA。随后,取7μL dscDNA用1.2% Agarose胶电泳检测。

表1 cDNA第二链合成反应体系
(2)cDNA与三框attB1重组接头连接(三种接头分别各连接一份)
在PCR管中依次加入4.5μL attB1 Adapter(1μg/μL)F、4.5μL attB1 Adapter(1μg/μL)R、1μL 10×NEB Buffer2,混匀后置于PCR仪中:95℃ 5min→25℃ 45min;向上步反应体系中加入34μL cDNA、5μL 10×Ligation Buffer、1μL T4 DNA Ligase(1U/μL),混匀后置于PCR仪中16℃反应24h。
(3)cDNA分级分离及收集
将93μL cDNA样品加入CHROMA SPINTM+TE-1000纯化柱内,多次颠倒使胶基质重悬并均匀;去除顶端的盖子和底部的塞子,将纯化柱置于2mL离心管中。在1000rpm条件下离心5min,让柱子处于半干状态,并将其转移至新的收集管中,然后向柱内再加入93μL cDNA,再次1000rpm离心5min,收集产物至新的2mL离心管中。向管内依次加入1/10体积的3M NaAc、2.5倍体积的无水乙醇,上下颠倒混合均匀后置于-20℃冰箱静置1h。置于离心机在4℃、14000rpm条件下离心25min,除去上清,保留沉淀。用1mL 70%乙醇溶液洗涤沉淀两次:4℃、14000rpm、3min,去除上清液。沉淀在室温条件下晾干后,再加入20μL DEPC water溶解cDNA,用于酵母文库的构建。
(4)BP重组反应
浓缩纯化后的双链cDNA与pDONR222载体进行BP重组反应,在PCR管内分别加入13μL dscDNA、2μL pDONR222载体(200ng/μL)、5μL Gateway®BP Clonase®II enzyme mix,混匀后置于PCR仪中25℃反应20h。
(5)转化
BP重组产物采用电转化(1500V,200Ω,25uF)方式转入大肠杆菌DH10B细胞内,加入4mLSOC培养液,置于37℃恒温振荡摇床250rpm培养1h,向菌液中加入无菌甘油至终浓度20%,即为酵母双杂交初级文库菌液,置于-80℃长期保存。
(6)库容量鉴定
将上步转化后的菌液吸取10μL,并加入ddH2O稀释100倍,取稀释后的菌液50μL涂布于LB固体培养基上(含Kana抗性),置于37℃恒温生化培养箱内过夜培养12~14h;次日,根据CFU/mL=平板上的克隆数/50μL×100倍×1000μL、文库总CFU=CFU/mL×文库菌液总体积mL,计算初级文库的库容量。
(7)重组率和插入片段长度鉴定
从LB菌落平板上随机挑取24各克隆进行菌落PCR鉴定,以pDONR222F、pDONR222R为引物(表2),采用2×Rapid Taq Master Mix进行PCR扩增,扩增产物用1% Agarose胶电泳检测。

表2 本实验中所用到的引物
1.2.4 安农1589穗部cDNA次级文库构建及质量鉴定
将上步cDNA初级文库菌液接种于肉汤培养基(含Kana抗性),30℃ 220rpm过夜振荡培养,次日采用FastPure Plasmid Mini Kit提取质粒。质粒测OD260后稀释至300ng/μL,在PCR管内依次加入1μL 初级文库质粒、1μL 次级文库载体(核体系为pGADT-DEST,膜体系为pPR3-N-DEST)(300ng/μL)、4μL GatewayTMLR ClonaseTMII enzyme mix、14μLddH2O,混匀后置于PCR仪中进行LR重组反应(25℃,20h)。重组产物采用电转化(1500V,200Ω,25uF)方式转入大肠杆菌DH10B细胞内,加入4mLSOC培养液,置于37℃恒温振荡摇床250rpm培养1h,向菌液中加入无菌甘油至终浓度20%,即为酵母双杂交次级文库菌液,置于-80℃长期保存。次级文库库容量鉴定及重组率和插入片段长度鉴定参照1.2.3(6)与1.2.3(7),核体系次级文库菌落PCR鉴定引物为pGADT-DEST-F、pGADT-DEST-R(表2),膜体系次级文库菌落PCR鉴定引物为pPR3-N-F、pPR3-N-R(表2),注意次级文库质粒为Amp抗性。
1.2.5 核体系cDNA次级文库质粒转化Y187酵母菌株
向无菌预冷的15mL离心管中依次加入600μL Y187酵母感受态细胞、5μg核体系次级文库质粒、20μL变性Carrier DNA(100℃ 5min,2次)、2.5mL PEG/LiAc溶液,轻轻涡旋震荡混合均匀,置于30℃恒温水浴锅孵育45min,每隔15min上下颠倒混合一次;再加入160μL DMSO,混合后置于42℃恒温水浴锅孵育20min,每隔10min上下颠倒混合一次。转化完成后,700g离心5min,移弃上清,并用30mL 0.9% NaCl溶液重悬细胞,涂布于SD/-Leu单缺培养基上,每板涂300μL 菌液,倒置于30℃恒温培养箱培养3~6d。
每块平板加5mL冷藏的培养基收集转化子,混合均匀后,取100μL经10000倍稀释后的菌液涂布于SD/-Leu单缺培养基上,倒置于30℃恒温培养箱培养2~3d,计算文库的滴度(滴度(cells/mL)=克隆数/涂板体积×稀释倍数)。从板中随机挑取24个单菌落于培养液中,250rpm过夜培养,次日10000rpm离心2min,除去上清。用200μL 0.2%SDS溶液重悬菌体,置于95℃条件下加热15min,低速离心后取上清作为PCR模板,以T7SP、3′AD为引物(表2),采用采用2×Rapid Taq Master Mix进行PCR扩增鉴定。
2 结果与分析
2.1 总RNA的提取、mRNA的分离纯化和dscDNA的合成
采用植物多酚多糖RNA提取试剂盒提取F.graminearum处理的小麦穗部不同时间点的总RNA,经1.5% Agarose胶电泳检测后主带清晰可见(图1A),28S RNA与18S RNA的亮度比接近2:1,无降解条带,表明RNA完整性好。通过微量核酸分析仪测定提取的总RNA样品的浓度在890.3~3079.4ng/μL,OD260/OD280在1.89~2.02,OD260/OD230在2.10~2.20,表明RNA纯度高,基本无其他杂质污染。
将F.graminearum处理不同时间的总RNA进行等比例混合,使用Oligotex mRNA Midi Kit分离纯化样本mRNA,经1%Agarose胶电泳显示mRNA条带清晰(图1B),在750~2500bp呈一条均匀集中的弥散壮状条带,表明分离的mRNA质量完好,无降解。
分离纯化的mRNA使用反转录试剂盒合成dscDNA,取部分产物经1%Agarose胶电泳检测。结果显示(图1C),dscDNA分布在100~2500bp,表明不同大小和丰度的mRNA均得到了有效的反转录,符合构建cDNA文库的需要。
2.2 接种F. graminearum的小麦穗部cDNA初级文库的构建和鉴定
按照CloneMinerTMII说明书进行构建cDNA初级文库,将反转录得到的dscDNA与三框attB1重组接头连接,再利用CHROMASPINTM+TE-1000对双链cDNA进行分级分离及收集和浓缩纯化,纯化后的dscDNA与pDONR222载体进行BP重组反应,最后将BP重组产物电转化到大肠杆菌DH10B细胞内,经培养后获得cDNA初级文库菌液。
从cDNA初级文库转化后大肠杆菌原液中吸取10μL,并稀释100倍,再从稀释后的菌液中取出50μL涂布于LB培养基(含Kana)上,倒置于37℃恒温培养箱过夜培养。次日对培养基进行cDNA文库库容量鉴定,经统计共长出1300个单菌落(图2A),计算得出初级文库库容量为2.6×106cfu/mL,总克隆数CFU为1.04×107。从培养基上随机挑取24个单菌落,以pDONR222F/pDONR222R为引物,使用2× Rapid Taq Master Mix进行菌落PCR鉴定,鉴定结果(图2B)显示插入片段分布在750~2000bp,平均长度大于1000bp,重组率100%。以上结果表明所构建的cDNA初级文库质量良好,可用于后续cDNA次级文库的构建。
2.3 接种F. graminearum的小麦穗部cDNA次级文库的构建和鉴定
从cDNA初级文库菌液中提取cDNA初级文库质粒,采用GatewayTMLR ClonaseTMII enzyme mix将初级文库质粒分别与pGADT-DEST载体(核体系)、pPR3-N-DEST载体(膜体系)进行LR重组反应。重组产物分别电转化到大肠杆菌DH10B细胞内,经培养后获得核体系cDNA次级文库菌液和膜体系cDNA次级文库菌液。从两种体系cDNA次级文库转化后大肠杆菌原液中各吸取10μL,并稀释100倍,再从稀释后的菌液中取出50μL分别涂布于LB培养基(含Amp)上,倒置于37℃恒温培养箱过夜培养。次日对培养基进行cDNA文库库容量鉴定,经统计核体系cDNA次级文库平板上共长出1500个单菌落(图3A),计算得出其文库库容量为3.0×106cfu/mL,总克隆数CFU为1.2×107;膜体系cDNA次级文库平板上共长出1400个单菌落(图3C),计算得出其文库库容量为2.8×106cfu/mL,总克隆数CFU为1.12×107。从两种cDNA次级文库培养基上各自随机挑取24个单菌落,分别以pGADT-DESTF/pGADT-DESTR(核体系)、pPR3-N-DESTF/pPR3-N-DESTR(膜体系)为引物,使用2×Rapid Taq Master Mix进行菌落PCR鉴定,鉴定结果(图3B、3D)显示两种次级文库插入片段分布均在750bp以上,平均长度均大于1000bp,重组率均为100%。以上结果表明所构建的核体系cDNA次级文库与膜体系cDNA次级文库质量均良好,基本可以满足后续核体系或膜体系酵母双杂交筛选互作蛋白对文库的要求。
2.4 核体系酵母双杂交次级文库转化酵母与鉴定
提取核体系cDNA次级文库质粒,并转化到Y187酵母菌株中,涂布于SD/-Leu培养基上,待平板出现克隆后收集转化子,取100μL稀释10000倍的菌液涂布于培养基平板上进行文库滴度测定,结果计算得出文库滴度为8.0×107cells/mL(图4A),满足核系统酵母双杂交对菌液细胞密度的要求(>3.0×107cells/mL)。随机提取24个单菌落进行菌液PCR鉴定,结果(图4B)显示插入片段均在750bp以上,重组率为100%,这与核体系cDNA次级文库鉴定结果一致。
3 讨论
利用酵母双杂交技术能够将蛋白与蛋白之间微弱的、瞬时的作用体现出来,具有高通量且灵敏度高的优点,因此该技术常被用来筛选酵母cDNA文库中互作蛋白和检测蛋白质之间是否互作的手段,对发掘植物中的新蛋白功能和研究蛋白质互作网络具有重要作用[18]。近年来,酵母双杂交技术被广泛应用在研究病原菌侵染植物和植物防御反应机制中。He等以赤霉病抗性位点Fhb1中的TaPFT基因为诱饵,通过核系统酵母双杂交技术从F.graminearum侵染的苏麦3号小麦穗部酵母文库中筛选到23个可能与之互作的蛋白,这些蛋白主要涉及泛素化过程、氧化还原过程、蛋白质磷酸化等生化反应[19]。Song等同样以TaPFT为诱饵蛋白,从酵母文库中筛选到16个可能与其互作的蛋白,并通过酵母双杂交回转实验和双分子荧光互补实验(BiFC)验证了TaPFT确实能够与正向参与小麦对赤霉病的抗性的小麦篦麻毒素B类凝集素(TaRBL)互作[20]。Luan等以大豆花叶病毒(Soybeanmosaicvirus,SMV)P3蛋白为诱饵蛋白,采用膜系统酵母双杂交技术从SMV侵染的大豆cDNA文库中筛选出51个相互作用的蛋白,其中超敏反应病变诱导蛋白(HRLI)被多次筛选到,通过BiFC证实了这种互作关系[21]。陈玉婷等采用酵母双杂交技术从构建的受青枯菌诱导的花生根本酵母文库中筛选到与抗青枯病AhRRS5互作的蛋白AhSBT1.6,并证明了后者参与花生抗青枯病[15]。
通过酵母双杂交系统大规模筛选候选互作蛋白需要以构建高质量的cDNA文库为重要前提[22]。目前构建cDNA文库多基于SMART技术,但在该技术中合成dscDNA的LD-PCR反应会优先扩增高丰度表达基因和短片段cDNA,这就会使低丰度表达基因及大片段cDNA容易丢失,从而降低文库的代表性[23]。然而,基于体外同源重组连接再转化细胞的Gateway技术能够很好地避免了PCR扩增造成的低丰度基因丢失和扩增过程个别碱基突变等对文库的影响。本研究就是采用Invitrogen公司的CloneMineTMII试剂盒,基于Gateway技术构建小麦酵母杂交cDNA文库。cDNA文库的构建具有器官、组织、时期及胁迫相应特异性,由于赤霉病的发病部位主要在小麦穗部,因此,本研究材料选用F.graminearum侵染后不同时间点的小麦穗部,确保了病原菌侵染条件下小麦穗部特异表达基因的最大化收集。cDNA文库的质量主要取决于RNA的完整性和文库库容[24]。本研究采用多糖多酚植物总RNA提取试剂盒从接种F.graminearum后不同时间点的小麦穗部提取总RNA,28S和18S两条带清晰可见,其亮度约呈2:1的比例,无降解,OD260/OD280与OD260/OD230约在2.0左右,表明RNA较为完整且无明显杂质存在,满足cDNA文库构建的要求。分离纯化后的mRNA经反转录合成dscDNA,电泳显示条带呈弥散状且均匀地分布在100~2500bp,表明dscDNA的丰度较高,这为构建高质量的cDNA文库奠定了基础。若要筛选出低丰度的互作蛋白就需要文库具有较高的库容。本研究所构建的小麦穗部受接种F.graminearum诱导的核体系酵母双杂交文库库容量为3.0×106cfu/mL,膜体系酵母双杂交文库库容量为2.8×106cfu/mL,两种文库的插入率均为100%,插入片段平均长度均大于1kb,故此这两种文库的覆盖度和完整性均较好,能够满足低丰度互作蛋白的筛选。
4 结论
本研究构建了小麦接种F.graminearum后穗部组织核系统酵母双杂交文库和膜系统酵母双杂交文库,经文库库容量、重组率和插入片段鉴定,两种文库质量均良好,能够满足酵母双杂交筛选互作蛋白的需求。本研究为进一步研究小麦与禾谷镰刀菌互作机制及解析相关抗病基因的抗性机制奠定了基础。
猜你喜欢
轻音乐(2022年11期)2022-11-22
湖南农业大学学报(自然科学版)(2022年2期)2022-05-11
作物学报(2021年11期)2021-08-31
阅读(低年级)(2021年4期)2021-06-15
猪业科学(2021年3期)2021-05-21
山东畜牧兽医(2021年6期)2021-01-11
幽默大师(2020年10期)2020-11-10
中华诗词(2019年1期)2019-11-14
民族音乐(2018年5期)2018-11-17
趣味(语文)(2018年7期)2018-06-26
