
基于密度泛函理论的普克鲁胺分子反应活性位点预测*
2023-11-28 13:08唐海飞
广州化工 2023年13期
唐海飞
(湘潭医卫职业技术学院,湖南 湘潭 411104)
新冠肺炎(COVID-19)疫情自出现以来,不断向全球蔓延,给全人类带来了严重威胁,是目前全球都面临的严重公共卫生事件。寻找抗击新冠病毒的有效药物成为全社会共同挑战。从小分子药物中发现抗新冠病毒药物也是重要的策略之一。普克鲁胺(proxalutamide)是我国苏州开拓药业开发出来的雄激素受体拮抗剂和降解剂(结构见图1),主要用于治疗前列腺癌[1-2]。因新冠肺炎患者存在有性别年龄差异[3-4],研究者发现,普克鲁胺用于治疗新冠肺炎时能有效降低男性门诊患者住院率[5],加速病毒清除速度[6],改善患者的肺损伤[7],对轻、中、重症新冠患者积极有效,有望成为国产“抗疫”利器。
由于缺少病例,我国学者对该药治疗新冠肺炎研究较少。国外对于该药治疗新冠肺炎的研究主要集中在其有效性研究方面[5-8]。关于普克鲁胺药物分子本身反应活性位点的相关研究较少,尚未发现运用量子化学手段对其在分子水平进行结构和反应位点的研究。通过量子化学手段在分子水平研究药物结构及性质,所得结果定量、准确,并可实现可视化,所得结论可为药物深入研究提供重要的参考价值,具有重要意义。
因此,本文以普克鲁胺药物分子为研究对象,利用量子化学软件对其进行计算分析,预测反应活性位点,为进一步理解普克鲁胺化学性质、作用机理、构效关系,以及推动普克鲁胺的结构改造和进一步开发利用提供理论参考。

图1 普克鲁胺分子结构Fig.1 The structures of proxalutamide
1 方 法
依据量子化学密度泛函理论,在B3LYP/6-311++G**方法水平下运用Gaussian 16程序对普克鲁胺分子进行结构优化,得到稳定结构及波函数文件,运用Multiwfn 3.8软件[9]对分子表面静电势[10-11](electrostatic potential,ESP)、平均局部离子化能(average local ionization energy,ALIE)[12-13]、前线分子轨道[14]、原子电荷[15]进行了计算分析。同时,基于概念密度泛函理论,也对简缩福井函数[16]等局部描述符和电负性、化学势、化学硬度、化学软度、亲电指数及亲核指数等全局描述符进行了计算分析。
2 结 果
2.1 Mulliken电荷
通过查阅Gaussian 16的分子结构优化文件可以的到除氢原子外的各原子的Mulliken电荷,结果见表1。通过对比表1的数据可知,氮原子及氧原子含有较多的负电荷,碳原子含有较多正电荷,这主要是它们的电负性不同所致。氮原子中,咪唑环中N(14)及N(16)所含负电荷较多;氧原子中,噁唑环O(31)、羰基O(32)含负电荷较多。表明这几处易发生亲电反应。三氟甲基C(9)正电荷在整个分子中最多,原因是其与三个电负性最强的氟原子相连;另外,羰基C(15)、C(18)正电荷也较多,这也是由于氧原子吸电子能力较强的缘故。表明这三处易发生亲核反应。这与赵智等[17]分析布洛芬分子分子波函数时情况相似。

表1 普克鲁胺分子中除氢原子外各个原子的Mulliken电荷Table 1 Mulliken charge of each atom (except hydrogen) in proxalutamide molecule

续表1
2.2 分子表面静电势
普克鲁胺分子表面静电势极大值点和极小值点见图2(蓝色为极小值,红色为极大值),静电势极值点编号及数值见表2。不同静电势数值在分子表面的分布面积见图3。

表2 普克鲁胺分子表面静电势极值点大小及编号Table 2 The value and number of the extreme points of the electrostatic potential on the surface of proxalutamide molecule
结合图2和表2可知,普克鲁胺在分子中O原子及N原子附近主要分布极小值点,这主要是由原子的电负性决定的,原子电的负性越强,表明吸电子能力越强,故呈现极小值点。标号为1的极小值点为整个分子最小的极值点,在氰基N(8)原子附近。同时,羰基O(32)、O(33)及N(28)附近也存在较小的极值点。表明这四处电子丰富,在发生静电吸引形成复合物时比较活泼,也可能易发生亲电反应。相反地,编号为16的极大值点在咪唑环C(15)附近,此处的静电势数值在整个分子中最大。此外,分子中嘧啶环H(38)附近编号为19的极大值点数值也较大。一般情况下,复合物的形成源于静电相互吸引,且偏向于静电势最大值和最小值相互靠近。综合考虑空间位阻,可以得出普克鲁胺在以氢键、卤键等形式形成复合物时,氰基N(8)、咪唑环C(15)、嘧啶环H(38)将发挥主要作用。这对研究普克鲁胺分子晶体堆积、普克鲁胺与受体结合时有重要帮助。这与唐海飞等[18]关于阿司匹林药物的相关研究类似。

图3 普克鲁胺不同静电势区间表面积分布图Fig.3 Surface area distribution of prokluamide in different electrostatic potential intervals
从图3可知,普克鲁胺分子表面的静电势值主要集中在-10~20 kcal/mol之间。而大于30 kcal/mol或小于-30 kcal/mol的分布区域面积虽小,却决定反应活性位点。结合表2和图2可知,该面积主要集中在氰基N(8)、咪唑环C(15)及嘧啶环H(38)附近,与我们之前的分析一致。
综上,静电势分析预测普克鲁胺分子中氰基N(8)、咪唑环C(15)及嘧啶环H(38)为反应活性位点,在以静电吸引为主的反应中发挥重要作用。
2.3 平均局部离子化能
平均局部离子化能指的是分子失去电子变成离子所需的能量。分子表面局部离子化能越小,表明此处电子平均能量越高,电子容易挣脱束缚,越容易发生亲电和自由基反应。平均局部离子化能可以弥补表面静电势在分析亲电反应位点中的不足。图4为普克鲁胺分子表面平均局部离子化能分布图。

图4 普克鲁胺分子表面平均局部离子化能分布图Fig.4 The average local ionization energy of proxalutamide
从图4中可以看出,噁唑环双键、N(28)周围及嘧啶环N(23)蓝色区域尤为明显,表明这几处易发生亲电反应。
2.4 前线分子轨道
前线分子轨道在反应过程发挥着重要作用。前线轨道包括最高占据轨道(highest occupied molecular orbital,HOMO)以及最低未占轨道(lowest unoccupied molecular orbital,LUMO),其中HOMO束缚电子能力较差,具有给电子性质[14]。前线轨道能隙差越小,表示电子越容易发生跃迁,反应活性越强。
普克鲁胺的前线轨道结构图见图5,其中蓝色表示波函数负相位,绿色表示波函数正相位。普克鲁量胺HOMO轨道能为-7.146 eV,LUMO轨道能量为-2.569 eV,能隙差为4.577 eV。普克鲁胺HOMO轨道位于噁唑环双键、N(28)及O(31)附近,表明此处易受亲电试剂攻击,发生亲电反应,与局部离子化能分析结果一致。而LUMO轨道主要分布苯环C(1)、C(2)、C(4)上,表明这些位置发生亲核反应的可能性稍大。这与常瑞等[19]关于N-羟乙酰神经氨酸分子结构性质的研究结论类似。
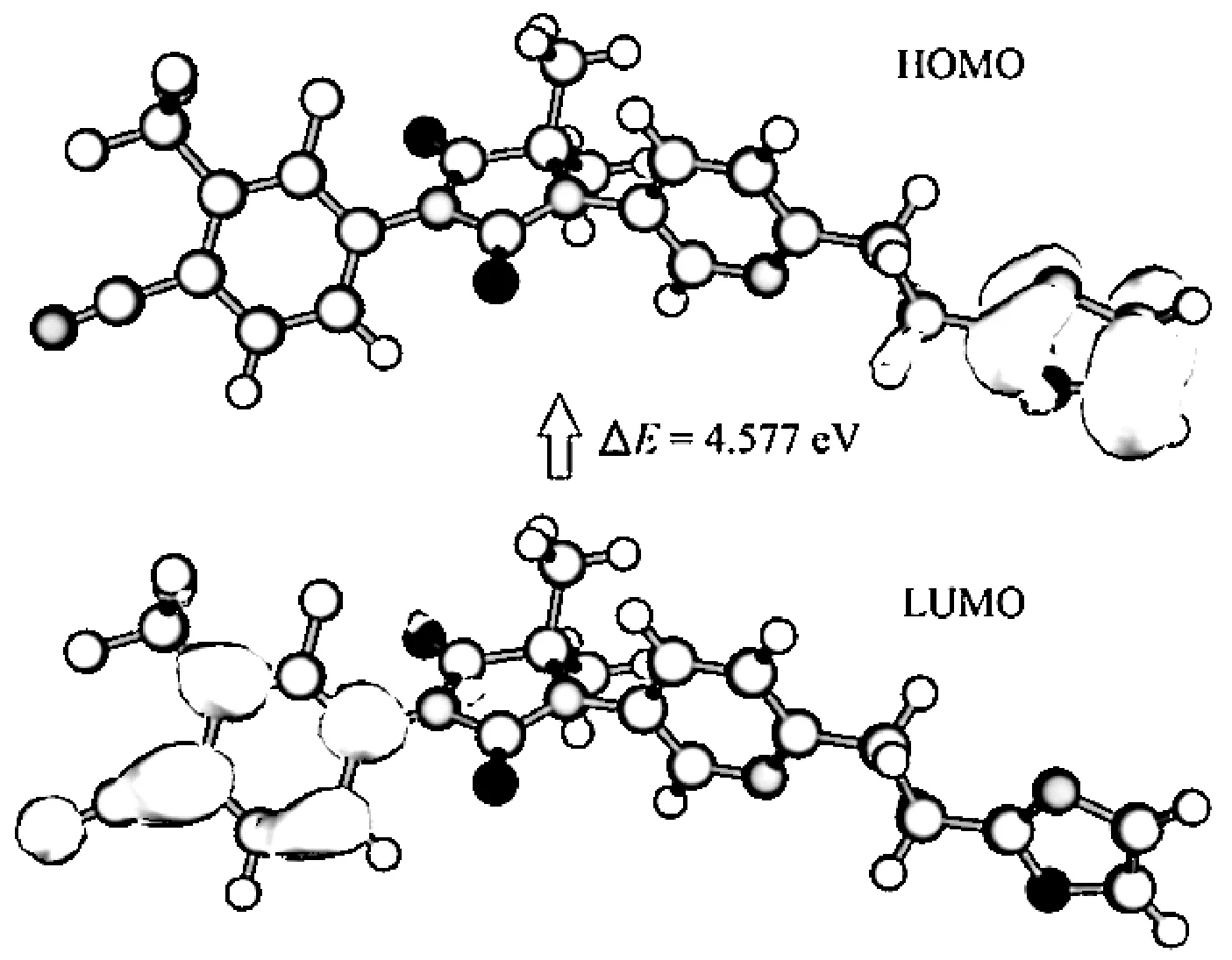
图5 普克鲁胺分子的HOMO和LUMO轨道图Fig.5 HOMO and LUMO orbital map of proxalutamide
2.5 原子电荷
原子偶极矩校正的Hirshfeld电荷(atomic dipole corrected Hirshfeld atomic charge,ADCH)可表征分子内不同原子所带电荷,带负电荷越多则亲电活性可能越强[15]。表3列出了普克鲁胺分子中较大及较小的ADCH电荷值。从表中可以看出,O(32)、O(33)、N(8)、N(28)具有较小原子电荷值,表明这几处具有较好的亲电活性。同时,C(9)、C(18)、H(38)、C(4)具有较大原子电荷值,表明这两个地方易发生亲核反应。这与前文分析所得结论类似,也与李怡菲等[20]研究环木菠萝烯醇阿魏酸酯分子结构性质所得结论类似。
2.6 简缩福井函数
简缩福井函数可定量分析分子中原子的亲电亲核反应活性,其中f+表示亲核活性,f-表示亲电活性[16]。由表4可知,普克鲁胺分子中,C(29)、C(30)、N(8)原子的f-值均较大,表明这三处易发生亲电反应。C(2)、C(5)、C(4)原子对应的f+值较大,可能是亲核反应活性位点。
全局活性参数也可用于预测化学反应活性。表5列出了普克鲁胺的全局活性参数,在比较普克鲁胺与其他药物的反应活性时可以进行参考。普克鲁胺亲电指数小,亲核指数大,表明更多的电子将从分子内转移出去,易被亲电试剂进攻,发生亲电反应。该分子硬度大,软度小,表明分子反应活性并不强。

表5 普克鲁胺的全局活性参数Table 5 Proxalutamide global acitivity paraeters
3 结 论
平均局部离子化能、前线轨道、简缩福井函数表明噁唑环双键为亲电反应位点,分子表面静电势、原子电荷、简缩福井函数表明氰基为亲电反应位点。前线轨道、原子电荷、简缩福井函数表明苯环C(4)为亲核反应活性位点。在以静电吸引为主的反应中,氰基N(8)、咪唑环C(15)、嘧啶环H(38)将发挥主要作用。本文全面分析了普克鲁胺分子的结构特征和反应活性位点,为进一步理解普克鲁胺化学性质、作用机理、构效关系,以及推动普克鲁胺的结构改造和进一步开发利用奠定了一定的理论基础。
猜你喜欢
广州化工(2022年19期)2022-11-09
广州化工(2022年18期)2022-10-22
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
分析化学(2020年8期)2020-08-21
硅酸盐通报(2020年1期)2020-02-25
分析化学(2018年4期)2018-11-02
分析化学(2018年7期)2018-09-17
低温与特气(2012年4期)2012-01-10
