高活性Cu-ZnO@SiO2纳米催化剂催化CO2加氢制甲醇
2023-11-24 08:01:16陈浩陈桂宋丹丹曾艳红刘文虎张明
高等学校化学学报 2023年11期
陈浩, 陈桂, 宋丹丹, 曾艳红, 刘文虎, 张明
高活性Cu-ZnO@SiO2纳米催化剂催化CO2加氢制甲醇
陈浩1, 陈桂2, 宋丹丹1, 曾艳红1, 刘文虎3, 张明4
(1. 岳阳职业技术学院生物环境工程学院, 岳阳 414000; 2. 怀化学院化学与材料工程学院, 怀化 418000; 3. 湖南石油化工职业技术学院石化工程学院, 4. 机电工程学院, 岳阳 414000)

催化剂; 二氧化碳加氢; 甲醇
CO2的大量排放造成了严重的环境问题(如全球变暖和气候变化), 但CO2同时也是一种宝贵的C1资源, CO2加氢制高附加值化学品(甲醇、 合成气和二甲醚)被认为是一种很有前景的CO2利用方式[1]. 其中, 甲醇不仅是一种重要的化工原料和优良的替代燃料, 它还可以进一步转化为甲醛、 醋酸、 二甲醚和各种石化产品, 因此, CO2加氢制甲醇被广泛关注[2,3]. 然而, CO2因其高稳定性和低反应活性而受到热力学限制, 同时, CO2加氢制甲醇的副反应——逆水煤气反应(RWGS)产生大量CO[4]. 因此, 需要开发高效的催化剂来提高CO2反应活性和甲醇选择性[5]. 已有文献[6~9]报道了贵金属(Pt, Pd, Au, Ag, Rh, Ru)和非贵金属(Cu, Ni)作为活性组分用于CO2加氢制甲醇. 众所周知, Cu基催化剂, 特别是 Cu/ZnO催化剂对甲醇具有较高的活性和选择性, 这是由于Cu-ZnO之间的强相互作用促进了Cu的分散和CO2的吸附, 从而具有较高的催化活性[10]. 另外, Zn向Cu表面的迁移产生了新的活性位点(如氧空位或铜-锌合金[10,11]). 然而, Cu/ZnO催化剂存在铜的烧结和催化剂容易失活等问题. 在工业甲醇合成过程中广泛应用的Cu/ZnO/Al2O3催化剂稳定性测试表明, 其活性仅为初始活性的2/3, 失活的主要原因是Cu纳米粒子的增长. SiO2具有很好的热稳定性, 是另一种优良的催化剂助剂, SiO2作为载体可以增加催化剂的表面积、 防止催化剂烧结, 从而提高催化活性和力学性能. 同时, 由于其表面羟基数量多, 可为金属物种提供丰富的位点, 因而催化剂分布均匀、 分散性好. 王继元等[12]研究了硅改性Cu-ZnO/HZSM催化剂对二甲醚(DME)合成的影响, 研究发现, SiO2可以抑制金属晶体的生长和聚集, CO2转化率更高. Jia等[13]发现, SiO2的添加明显改善了催化剂的BET表面积和铜的表面积, 加入4%(质量分数)SiO2的CuO-ZnO-ZrO2催化剂的CO2转化率为22.8%, 甲醇选择性提高到了45%. Wu等[14]认为SiO2改性有助于提高CuO/ZnO/ZrO2/Al2O3催化剂的稳定性. Phongamwong等[15]的研究表明, 在CuO-ZnO-ZrO2催化剂中加入1%的SiO2, 甲醇合成活性将提升26%. 然而, 大多数研究只关注催化活性与甲醇收率, 目前对于CO2加氢合成甲醇和CO的机理尚缺乏了解[16].
本文通过Stöber法合成二氧化硅纳米颗粒, 通过严格控制水的加入量从而控制颗粒大小, 将其作为载体合成Cu-ZnO@SiO2催化剂, 并用于CO2加氢制甲醇反应. 研究发现, SiO2可以提高金属Cu的分散性, 提高Cu+物种的比例, 进而提高甲醇的选择性和催化性能, 原位红外光谱表征分析表明, Cu-ZnO@SiO2催化剂具有与Cu-ZnO催化剂不同的催化反应路径.
1 实验部分
1.1 试剂与仪器
六合水硝酸锌[Zn(NO3)2·6H2O, 分析纯]、 三合水硝酸铜[Cu(NO3)2·3H2O, 分析纯]、 碳酸钠(Na2CO3, 分析纯)、 正硅酸四乙酯(TEOS, 分析纯)和氨水(NH3·H2O, 分析纯, 质量分数28.0%~30.0%), 购于国药集团化学试剂有限公司.
Rigaku D/Max-2550 VB+18 kW型X射线衍射仪(XRD), 日本理学公司; PLUS HD88/ASAP 2020型氮气吸附-脱附仪(BET), 美国麦克公司; ChemBET-3000型化学吸附仪[氢气程序升温还原(H2-TPR)和二氧化碳程序升温脱附(CO2-TPD)], 美国康塔公司; Thermo Scientific K-Alpha 1063型X射线光电子能谱仪(XPS)和Thermo fisher Nicolet iS10型原位红外光谱分析仪(DRIFTS), 美国赛默飞世尔科技公司; JEOL JSM-6610LV型扫描电子显微镜(SEM), 日本电子公司; Tecnai G2 F3型透射电子显微镜(TEM), 美国FEI公司.
1.2 实验过程
1.2.1催化剂的制备采用共沉淀法制备Cu-ZnO催化剂. 在烧杯中加入50 mL去离子水, 然后在70 ℃ 搅拌下, 将2.98 g Cu(NO3)2·3H2O和3.66 g Zn(NO3)2·6H2O的30 mL混合溶液和沉淀剂Na2CO3溶液(1.0 mol/L)同时滴加到50 mL去离子水中, 沉淀过程中控制pH恒定为7.0, 滴加完成后继续搅拌 1 h, 室温过夜老化, 产物用大量去离子水洗涤以去除钠离子, 固体产物于100 ℃干燥8 h, 然后在马弗炉中于350 ℃下煅烧5 h, 得到的催化剂前体标记为CuO-ZnO, 催化剂前体在10% H2⁃N2氛围下于300 ℃还原2 h, 得到的催化剂命名为Cu-ZnO.
Cu-ZnO@SiO2催化剂的制备. 首先, 用Stöber法合成二氧化硅微球胶体, 将正硅酸四乙酯(TEOS, 1.86 g)溶解在40 mL乙醇中, 然后加入4 mL氨水(28.0%~30.0%), 将得到的混合物在室温下搅拌1 h后, 加入若干体积的去离子水继续搅拌1 h. 配制Cu(NO3)2·3H2O(0.27 g)和Zn(NO3)4·6H2O(0.33 g)的乙醇溶液20 mL, 然后迅速加入SiO2胶体中, 在室温下继续搅拌12 h, 离心, 用乙醇洗涤3次, 得到的产物在60 ℃下真空干燥过夜, 置于马弗炉中于350 ℃下煅烧5 h, 将得到的催化剂前体标记为 CuO-ZnO@SiO2, 催化剂前体在10% H2⁃N2氛围下于300 ℃还原2 h, 得到的催化剂命名为Cu-ZnO@SiO2.



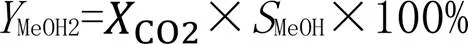
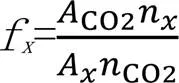
式中:A为各物质的峰面积;n(mol)为各物质的摩尔数;f为各物质相对CO2的校正因子.
2 结果与讨论
2.1 样品的表征
图1为CuO-ZnO和CuO-ZnO@SiO2催化剂的XRD谱图, 2=25°处谱峰可归属于非晶体的硅, 2=31.7°, 34.4°和36.2°处谱峰可归属于六方纤锌矿ZnO(JCPDS No.36-1451)(100), (002)和(101)晶面的特征衍射峰. 2=35.5°和38.8°处谱峰可归属于单斜型CuO(JCPDS No.45-0937)(002), (111)晶面的特征衍射峰. 在CuO-ZnO催化剂中, 2=35.5°的CuO(002)晶面和2=34.4°, 36.25°的ZnO(002), (101)晶面存在部分重叠, CuO-ZnO@SiO2催化剂中未观察到明显的CuO和ZnO的衍射峰, 这表明CuO和ZnO在CuO-ZnO@SiO2催化剂中的高度分散.
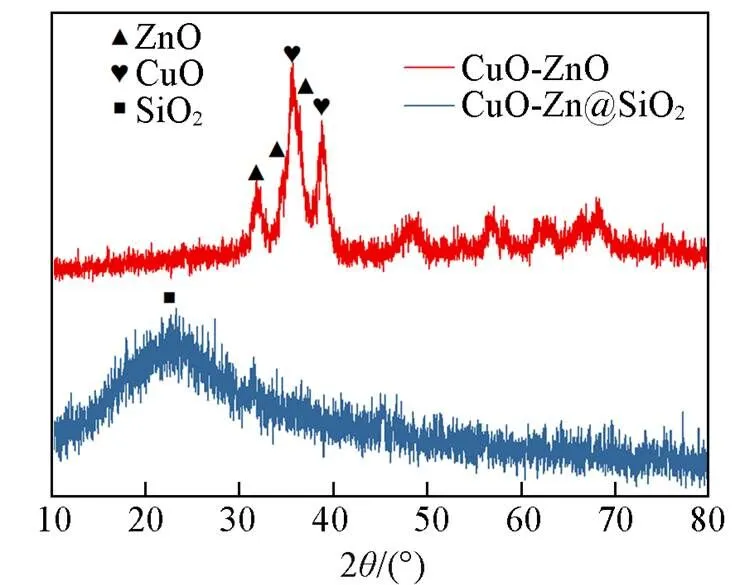
Fig.1 XRD patterns of CuO⁃ZnO and CuO⁃ZnO@SiO2 catalysts
通过氮气吸附-脱附分析了CuO-ZnO和CuO-ZnO@SiO2催化剂的物理性质, 结果如图2所示. 所有等温线[图2(A)]均为V型, 滞后环为H型, 催化剂具有介孔结构. 孔径分布曲线如图2(B)所示, CuO-ZnO催化剂孔径在5~60 nm范围内分布很宽泛, 而CuO-ZnO@SiO2催化剂的孔径分布更集中, 为 0~30 nm. BET比表面积(BET)、 孔体积、 平均孔径数据列于表1, CuO-ZnO催化剂的比表面积和孔容分别为58.5 m2/g和0.10 cm3/g, CuO-ZnO@SiO2催化剂具有更大的比表面积(80.2 m2/g)和孔容(0.13 cm3/g), 孔径(10.2 nm)相比CuO-ZnO催化剂(25.9 nm)更小. 这表明CuO-ZnO催化剂为大颗粒形成的堆积孔, 而CuO-ZnO@SiO2催化剂具有丰富的孔道和高的比表面积, 有助于防止催化剂的烧结[17].
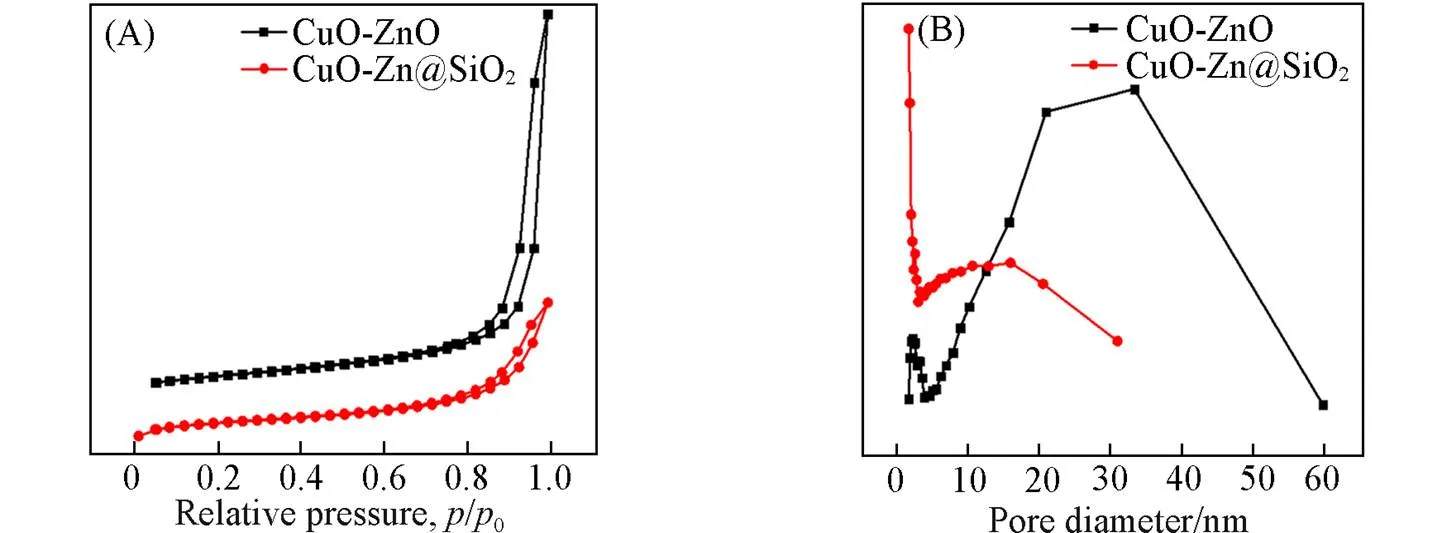
Fig.2 Adsorption⁃desorption isotherms(A) and pore size distributions(B) of the CuO⁃ZnO and CuO⁃ZnO@SiO2 catalysts

Table 1 Textural properties of the CuO-ZnO and CuO-ZnO@SiO2 catalysts
图3(A)和(B)分别为CuO-ZnO和CuO-ZnO@SiO2催化剂的SEM照片, 通过对比可以发现, 两种催化剂均呈均匀分散. 通过TEM照片进一步分析催化剂的粒径, 可以看出, CuO-ZnO催化剂颗粒呈不规则形状[图4(A)], 通过Nanomeasureer 1.2软件分析颗粒粒径约为14 nm[图4(B)]. 图4(C)为 CuO-ZnO@SiO2催化剂的TEM照片, 图中催化剂大部分呈球形颗粒, 粒径约为37 nm[图4(D)], 由于采用Stöber法合成二氧化硅微球, 通过控制正硅酸乙酯的水解速率, 可以调节二氧化硅颗粒的大小, 通常制备的二氧化硅微球一般在200 nm左右, 通过严格控制制备过程中水的加入量, 能有效防止微球的聚集增大, 从而制备小颗粒二氧化硅微球; 进一步观察微球表面, 可以看到, 微球表面有大量分散均匀的小颗粒[图4(E)], 大小在2.5 nm左右[图4(F)]. 图4(G)为高分辨透射电子显微镜(HRTEM)照片, 图4(H)为局部放大图, 在部分颗粒上可以观察到晶格间距为0.23 nm、 可归属于CuO的(111)晶面, 这表明微球表面的小颗粒物质为CuO, 同时, 并未发现ZnO的晶格, 说明ZnO在催化剂表面呈高度分散状.

Fig.3 SEM images of CuO⁃ZnO(A) and CuO⁃ZnO@SiO2(B) catalysts

Fig.4 TEM images(A, C, E) and particle size distribution histograms(B, D, F) and HRTEM image(G) of the CuO⁃ZnO(A, B) and CuO⁃ZnO@SiO2(C—H) catalysts
对催化剂进行H2-TPR实验, 以评估SiO2的加入是否对铜的还原性有影响. 图5为催化剂经10% H2⁃N2还原程序得到的H2-TPR谱图. 从图中可以看到, 两种催化剂在50~800 ℃的温度范围内均出现明显的还原峰, 可归属于CuO的还原峰, 对于负载催化剂, 普遍认为高分散CuO的还原峰一般位于 240~280 ℃区域[18]. CuO-ZnO催化剂还原峰温度为317 ℃, 而CuO-ZnO@SiO2催化剂还原峰温度更低, 只有268 ℃, 这表明SiO2的加入极大地促进了CuO的分散, 从而降低了催化剂的还原温度.

Fig.5 H2⁃TPR profiles of the CuO⁃ZnO and CuO⁃ZnO@SiO2 catalysts
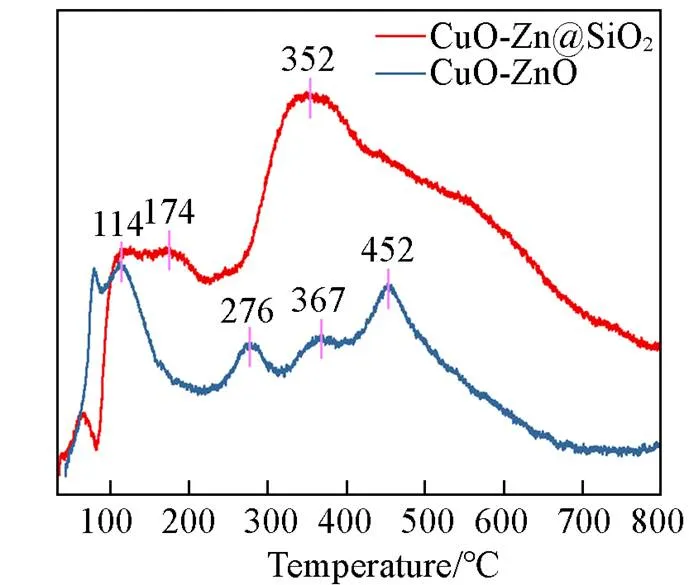
Fig.6 CO2⁃TPD profiles of the Cu⁃ZnO and Cu⁃ZnO@SiO2 catalysts
图6为Cu-ZnO和Cu-ZnO@SiO2催化剂的CO2-TPD谱图, 在50~200 ℃温度范围内的低温解吸峰为弱碱性位点, 可归属于表面羟基. 200~600 ℃为高温解吸峰, 代表强碱性位点, 与金属-氧对(Zn-O)和配位不饱和的O2-离子(低配位氧原子)有关. Gao等[19]认为CO2加氢通过甲酸盐路径生成中间物种甲醛(H2CO), H2CO加氢生成甲醇, 解离生成副产物CO, 强碱性位点更有利于促进CO2加氢生成甲醇. 从 图6可以看出, 与Cu-ZnO催化剂相比, Cu-ZnO@SiO2催化剂的低温解吸峰和高温解吸峰均有较大增加, 表明Cu-ZnO@SiO2催化剂拥有更多的碱性位点, 碱性位点的增加有利于CO2的吸附. 研究发现, 由于SiO2的碱度较低, SiO2表面吸附CO2的量要低于ZnO, 因此, SiO2的加入提高了CO2的吸附, 这可能归因于SiO2改善了活性组分的分散, 并导致活性组分与CO2之间的良好接触[15].
催化剂在进行XPS表征前先进行预处理, 在10% H2⁃N2氛围下于300 ℃还原2 h, 图7为Cu-ZnO和Cu-ZnO@SiO2催化剂的Cu2p, Zn2p, CuAuger和O1s的XPS谱图, 从图7(A)可以看出, Cu-ZnO催化剂中 Cu2p3/2的结合能为932.28 eV, Cu-ZnO@SiO2催化剂Cu2p3/2的结合能为932.58 eV, 移动了0.3 eV. 同时, 图7(B)中Zn2p的谱图也观察到类似的移动, Cu-ZnO催化剂中Zn2p3/2的结合能为1021.48 eV, 而 Cu-ZnO@SiO2催化剂Zn2p3/2的结合能为1022.38 eV, 结合能移动了0.9 eV. 这表明Cu/ZnO催化剂嵌入SiO2中, 新的Cu-Si和Zn-Si界面与Cu—O—Si和Zn—O—Si键形成, 较强的Si—O键使得电子更难从 Cu—O和Zn—O键中逃逸, 从而导致Cu和Zn的结合能增大[20]. 进一步分析催化剂表面Cu物种的价态, 由图7(A)中Cu2p曲线可以看出, 在941~945 eV之间未发现Cu2+卫星峰的存在, 表明催化剂表面只存在Cu+或/和Cu0物种, Cu-ZnO和Cu-ZnO@SiO2催化剂的CuAuger如图7(C)所示, Cu-ZnO催化剂的 Cu LMM曲线分解为2个峰, 916.6 eV处的峰归属于Cu+, 918.8 eV处的峰归属于Cu0, 以峰面积比值得到Cu+/Cu0摩尔比为2.8. CuO-ZnO@SiO2催化剂Cu+结合能位置移动到916.3 eV, Cu0结合能位置为918.6 eV, 此时Cu+/Cu0摩尔比为7.5, 这表明Cu-ZnO@SiO2催化剂中具有更高比例的Cu+物种, 这可归因于SiO2与Cu形成的Cu—O—Si键. Si等[21]认为Cu+位点主要吸附甲氧基和酰基, 而Cu0位点促进H2的解离, Liu等[22]的研究表明, CO加氢和CO2加氢均可合成甲醇, 当表层以Cu+为主时, CO是主要的碳源, 当金属Cu覆盖表面时, CO2是主要碳源, 在反应条件下, H2和CO容易将Cu2O还原为金属Cu. 而SiO2的加入能稳定Cu+. 以上结果表明, SiO2的加入促进了Cu+物种的形成和稳定, Cu+物种的存在将有利于副产物CO的吸附和转化为甲醇.

Fig.7 Cu2p(A), Zn2p(B), CuAuger(C) and O1s(D) XPS spectra of Cu⁃ZnO and Cu⁃ZnO@SiO2 catalysts
图7(D)为O1s谱图, Cu-ZnO的O1s曲线可以分解为两个峰, 以530.4和531.8 eV结合能为中心的晶格氧(OL)和表面氧空位(OV)分别来自纤锌矿结构中的晶格氧阴离子(O2-)和ZnO表面氧空位引起的缺氧区O-或O2-离子[23]. 由于SiO2富含表面羟基, 因此, Cu-ZnO@SiO2催化剂O1s曲线分解为以531.8和532.9 eV结合能为中心的OV和表面化学吸附氧(OC), OL的消失表明ZnO的高度分散或Zn以Zn—O—Si键的形式存在.
2.2 催化性能

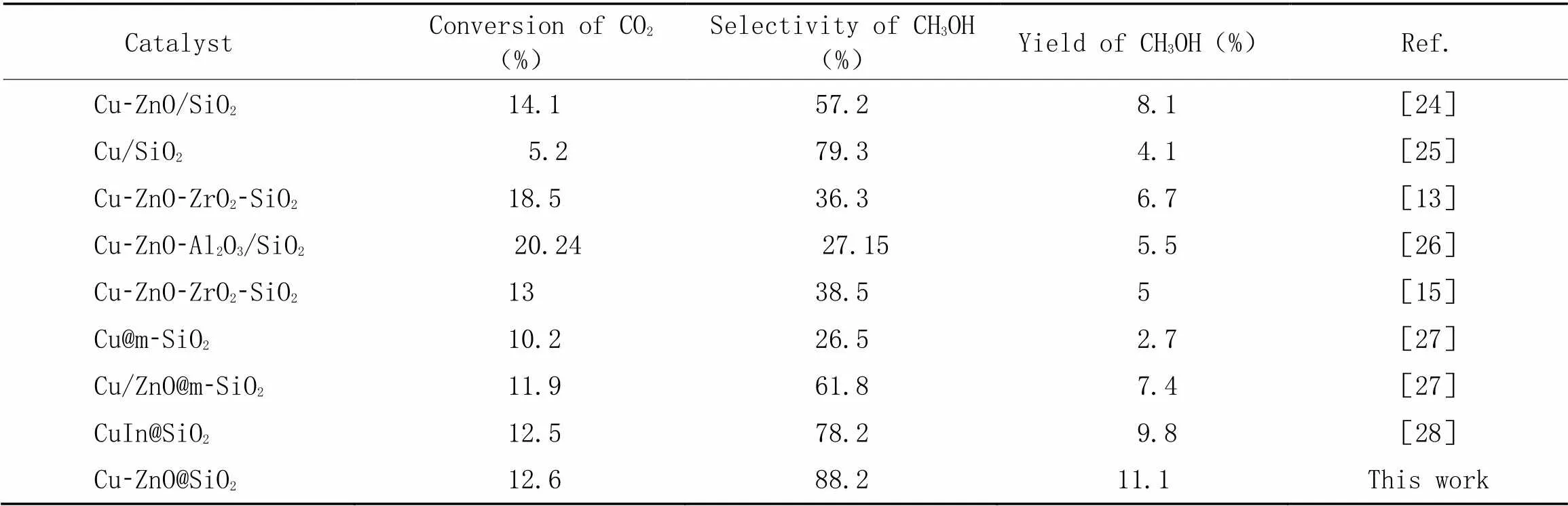
Table 2 Catalytic performance of Cu-based catalyst modified by SiO2 in CO2 hydrogenation to methanol
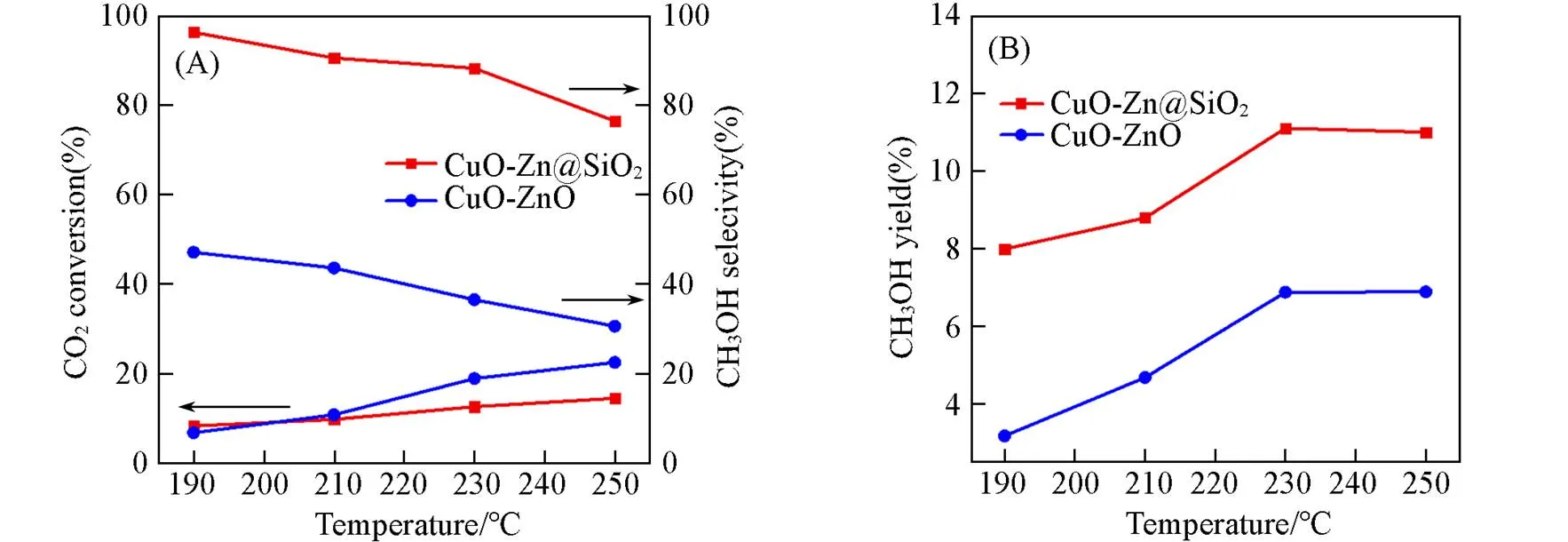
Fig.8 CO2 conversion, methanol selectivity(A) and methanol yield(B) of the Cu⁃ZnO and Cu⁃ZnO@SiO2 catalysts
2.3 反应机理
重点研究了CO2在Cu-ZnO和Cu-ZnO@SiO2催化剂上加氢制甲醇的反应机理, 相比之下, 目前对CO2在SiO2负载铜基催化剂上加氢制甲醇的反应机理了解较少, 为了研究反应路径, 在0.1 MPa, 230 ℃条件下, 通过原位DRIFTS光谱分析了CO2加氢制甲醇过程的中间物种, 结果如图9所示, 曲线的峰及其对应的化学物种列于表3. 高波数段(3500~3800 cm-1)为物理吸附CO2[29], 1102 cm-1为C—O共振, 可归属于甲氧基(CH3O*), 1596和1375 cm-1处的波峰可属于as(OCO)和s(OCO)的吸附峰[30], 2938, 2989 cm-1分别为(CH)和(CH)的吸附峰, 可均归属于甲酸盐物种[31], 在Cu-ZnO@SiO2催化剂中, 观察到CO*的吸收峰(2084 cm-1), 可归属于CO在Cu+物种上的吸附[32,15], 这与图7(C)中在 Cu-ZnO@SiO2催化剂中Cu+含量较高的结果一致, 同时, 并没有观察到甲酸盐的吸收峰, 而Cu-ZnO催化剂的结果却正好相反, 这表明CO2在Cu-ZnO催化剂中主要通过甲酸盐路径加氢生成甲醇, 而在 Cu-ZnO@SiO2催化剂上主要以RWGS(逆水煤气反应)+CO加氢路径进行[30]. CO2加氢制甲醇的反应机理如Scheme 1所示, 氢气在Cu金属表面解离, ZnO表面吸附的CO2通过RWGS反应生成CO, Cu-ZnO界面吸附的CO2通过甲酸盐路径生成CH3OH. Yu等[25]认为Cu+抑制了CO*中间物种的解吸, 从而抑制CO的生成, 并通过RWGS+CO加氢路径进一步生成CH3OH. 图4的TEM照片显示, SiO2表面的铜物种具有更高的分散性, 图7(C)的XPS谱图表明, Cu-ZnO@SiO2催化剂具有更高比例的Cu+含量.

Table 3 IR peak assignments of intermediate species over catalyst surface in CO2 hydrogenation
3 结 论
采用Stöber法合成了二氧化硅纳米颗粒, 作为载体制备了Cu-ZnO@SiO2催化剂并用于CO2加氢制甲醇, 并与传统共沉淀法制备的Cu-ZnO催化剂进行了对比. 结果表明, Cu-ZnO@SiO2催化剂具有更高的Cu分散性和更高比例的Cu+物种, Cu+抑制了CO*中间物种的解吸附, 从而抑制CO的生成. 因此, 相比Cu-ZnO催化剂(甲醇选择性36.5%, 甲醇收率6.9%), Cu-ZnO@SiO2催化剂具有更高甲醇选择性(88.2%)和甲醇收率(11.1%). 研究结果表明, Cu在SiO2表面形成的Cu—O—Si键是其Cu+含量较高的原因, DRIFT表征证实CO2在Cu-ZnO催化剂上主要通过甲酸盐(HCOO*)路径加氢生成甲醇, 而在 Cu-ZnO@SiO2催化剂上主要通过RWGS+CO加氢路径生成甲醇.
[1] Wang H., Fan S., Wang S., Dong M., Qin Z. F., Bin Fan W., Wang J. G.,., 2021,(11), 1609—1619
[2] Álvarez A., Bansode A., Urakawa A., Bavykina A. V., Wezendonk T. A., Makkee M., Gascon J., Kapteijn F.,., 2017,(14), 9804—9838
[3] Ye R. P., Ding J., Gong W., Argyle M. D., Zhong Q., Wang Y., Russell C. K., Xu Z., Russell A. G., Li Q., Fan M., Yao Y. G.,, 2019,(1), 1—15
[4] Kondratenko E. V., Mul G., Baltrusaitis J., Larrazábal G. O., Pérez⁃Ramírez J.,, 2013,(11), 3112—3135
[5] Wang Y. Y., Liu H. Z., Han B. X.,, 2020,(11), 2393—2403(王艳燕, 刘会贞, 韩布兴. 高等学校化学学报, 2020,(11), 2393-2403)
[6] Din I. U., Shaharun M. S., Alotaibi M. A., Alharthi A. I., Naeem A., J. CO Util., 2019,, 20—33
[7] Dang S., Yang H., Gao P., Wang H., Li X., Wei W., Sun Y.,, 2019,, 61—75
[8] Zhong J., Yang X., Wu Z., Liang B., Huang Y., Zhang T.,, 2020,(5), 1385—1413
[9] Zhou L. L., Cheng H. Y., Zhao F. Y.,, 2022,(7), 20220279(周雷雷, 程海洋, 赵凤玉. 高等学校化学学报, 2022,(7), 20220279)
[10] Rodriguez J. A., Liu P., Stacchiola D. J., Senanayake S. D., White M. G., Chen J. G.,., 2015,(11), 6696—6706
[11] Jiang X., Nie X., Guo X., Song C., Chen J. G.,., 2020,(15), 7984—8034
[12] Wang J. Y., Zeng C. Y., Wu C. Z.,., 2006,(2), 195—199(王继元, 曾崇余, 吴昌子, 燃料化学学报(中英文), 2006,(2), 195—199)
[13] Jia M. Y., Gao W. G., Wang H., Wang Y. H.,., 2014,—, 117—122
[14] Wu J., Luo S., Toyir J., Saito M., Takeuchi M., Watanabe T.,, 1998,(1—4), 215—220
[15] Phongamwong T., Chantaprasertporn U., Witoon T., Numpilai T., Poo⁃arporn Y., Limphirat W., Donphai W., Dittanet P., Chareonpa⁃nich M., Limtrakul J.,, 2017,, 692—703
[16] Jia C. X., Shao J. A., Bai I. X. W., Xiao J. J., Yang H. P., Chen H. P.,., 2020,(9), 3658—3668(贾晨喜, 邵敬爱, 白小薇, 肖建军, 杨海平, 陈汉平, 化工进展. 2020,(9), 3658-3668)
[17] Dai W. H., Xin Z.,., 2022,(8), 3586—3596(戴文华, 辛忠, 化工学报. 2022,(8), 3586—3596)
[18] Chen K., Yu J., Liu B., Si C., Ban H., Cai W., Li C., Li Z., Fujimoto K.,, 2019,, 163—173
[19] Gao P., Li F., Zhang L., Zhao N., Xiao F., Wei W., Zhong L., Sun Y.,J. CO Util., 2013,, 16—23
[20] Cui X., Yan W., Yang H., Shi Y., Xue Y., Zhang H., Niu Y., Fan W., Deng T.,., 2021,, 2661—2672
[21] Si C., Ban H., Chen K., Wang X., Cao R., Yi Q., Qin Z., Shi L., Li Z., Cai W., Li C.,:., 2020,, 117466
[22] Liu Y. M., Liu J. T., Liu S. Z., Li J., Gao Z. H., Zuo Z. J.,J. CO Util., 2017,, 59—65
[23] Huang C., Wen J., Sun Y., Zhang M., Bao Y., Zhang Y., Liang L., Fu M., Wu J., Ye D., Chen L.,, 2019,, 221—230
[24] Jiang Y., Yang H., Gao P., Li X., Zhang J., Liu H., Wang H., Wei W., Sun Y., J. CO Util., 2018,, 642—651
[25] Yu J., Yang M., Zhang J., Ge Q., Zimina A., Pruessmann T., Zheng L., Grunwaldt J. D., Sun J.,., 2020,(24), 14694—14706
[26] Zhang L., Zhang Y., Chen S.,, 2012,, 118—123
[27] Yang H., Gao P., Zhang C., Zhong L., Li X., Wang S., Wang H., Wei W., Sun Y.,., 2016,, 56—60
[28] Shi Z., Tan Q., Wu D.,., 2019,(3), 1047—1058
[29] Shi Z., Tan Q., Tian C., Pan Y., Sun X., Zhang J., Wu D.,., 2019,, 78—89
[30] Wang S. Q., Yang J. H., Zhao N., Xiao F. K.,, 2022,(7), 1—7(王世强, 杨金海, 赵宁, 肖福魁. 燃料化学学报(中英文), 2022,(7), 1—7)
[31] Song J., Liu S., Yang C., Wang G., Tian H., jian Zhao Z., Mu R., Gong J.,:, 2020,, 118367
[32] Liu T., Hong X., Liu G.,., 2020,(1), 93—102
High-performance Cu-ZnO@SiO2Nano-catalyst for CO2Hydrogenation to Methanol
CHENHao1, CHENGui2, SONGDandan1, ZENGYanhong1, LIUWenhu3*, ZHANGMing4*
(,,414000,;,,418000,;,,,414000,)
Catalyst; CO2hydrogenation; Methanol
2023-06-05
刘文虎, 男, 硕士, 讲师, 主要从事C1资源转化方面的研究. E⁃mail: liuwenhu2023@163.com
张 明, 男, 学士, 讲师, 主要从事C1资源转化方面的研究. E-mail: ming84122023@163.com
湖南省自然科学基金(批准号: 2023JJ50309,2023JJ50450)资助.
O643.36; X773
A
10.7503/cjcu20230268
2023-08-04.
Supported by the Natural Science Foundation of Hunan Province, China(Nos.2023JJ50309,2023JJ50450).
(Ed.: V, K, S)
猜你喜欢
化工管理(2022年14期)2022-12-02 11:46:54
大学物理(2022年9期)2022-09-28 01:10:52
潍坊学院学报(2020年6期)2020-11-22 08:04:10
物理通报(2020年7期)2020-07-01 09:28:02
石油石化绿色低碳(2019年6期)2019-02-13 09:39:01
中国化肥信息(2016年27期)2016-05-17 04:25:18
大连工业大学学报(2015年4期)2015-12-11 04:06:50
原子与分子物理学报(2015年3期)2015-11-24 12:49:34
文理导航(2015年26期)2015-09-29 14:12:24
中国当代医药(2015年29期)2015-03-01 02:07:41