
端羧基超支化聚酰胺-胺的制备及阻垢性能
2023-11-15 03:57王洋洋刘庆旺范振忠付沅峰仝其雷
石油化工 2023年10期
王洋洋,刘庆旺,范振忠,付沅峰,仝其雷
(东北石油大学 石油工程学院 提高采收率教育部重点实验室,黑龙江 大庆 163188)
在三次采油中,由于注入流体的不相容性、热 力学条件的变化,导致采出水因过度饱和而产生大面积结垢[1-2],并且随着时间的推移这些垢质会堵塞近井眼或油井孔吼,导致地层孔隙度和渗透率降低,为了克服这些问题,最具成本效益和广泛采用的解决方案之一是加入阻垢剂[3]。超支化聚合物是近些年来兴起的一种高效绿色阻垢剂,由于具有三维网状结构、超低密度表面官能团和末端基易修饰改性等特点[3-4],在制备油田防垢、阻垢剂方面具有十分重要的发展前景。Yao 等[4]采用2-磷酸基-1,2,4-三羧酸丁烷和琥珀酸酐分别对1.0 ~2.0 代的超支化聚酰胺-胺(PAMAM)大分子进行末端基改性,发现改性超支化大分子不仅阻垢性能增强,而且在Ca2+含量为4 000 mg/L 和pH=5 ~9时,仍可抑制85%以上的水垢。李美兰等[5]以衣康酸、三乙醇胺和琥珀酸为单体,通过酯化反应制备出一种具有生物降解性能的无磷型绿色端羧基超支化聚酯,通过静态阻垢性能测试,发现它对钙垢的阻垢率达90%以上,生物降解率在28 d时达到67.2%。苏高申等[6]采用氯乙酸作改性剂制备了1.0 代和2.0 代端羧基超支化PAMAM 阻垢剂,可有效解决中国石油长庆油田华子坪区块存在的CaCO3,CaSO4垢问题。
21 世纪初,随着点击化学中铜催化叠氮-炔烃环加成和硫醇-烯/炔加成反应在超支化聚合物合成方面的应用[7-9],使得该类聚合物在阻垢防垢方面的应用更多样化。马来酸酐(MAH)共聚物阻垢剂被认为是无毒、污染性小和环境可接受的水处理药剂[10],但也存在钙容忍度低、聚合度低等问题,因此在MAH 共聚物中添加超支化分子结构,可有效提高它的分子量、阻垢性能和抗矿化度能力。
本工作以多乙烯多胺为反应的中心核,MAH为封端剂,通过Michael 加成和酰胺化反应制备了端羧基超支化PAMAM 阻垢剂。利用FTIR、元素分析、1H NMR、TG、UV-Vis、SEM 等方法对聚合物结构进行了表征,考察了不同条件对端羧基超支化PAMAM 阻垢性能的影响,并推测了阻垢机理,为研究超支化聚合物阻垢剂的现场应用提供理论依据。
1 实验部分
1.1 主要试剂和仪器
乙二胺(EDA)、二乙烯三胺(DETA)、三乙烯四胺(TETA)、丙烯酸甲酯、MAH:分析纯,上海麦克林生化科技有限公司;丙酮:分析纯,西陇科学股份有限公司;氯化钙:分析纯,福晨(天津)化学试剂有限公司;甲醇:分析纯,天津市凯通化学试剂有限公司;FeSO4·7H2O:分析纯,天津市科密欧化学试剂有限公司。
Nicolet IS50 型傅里叶变换红外光谱仪:赛默飞世尔科技公司;PE2400 Ⅱ型元素分析仪:珀金埃尔默公司;Bruter 600M 型核磁共振波谱仪:Varian 公司;TGA-1000 型热重分析仪:上海群弘仪器设备有限公司;721 型紫外-可见分光光度计:上海佑科仪器仪表有限公司;Nano ZS 型Zeta 电位仪:马尔文仪器有限公司;JSM7200F 型扫描电子显微镜:日本电子株式会社。
1.2 试样制备
将质量比1∶4 的EDA 和甲醇的混合溶液加入带有回流冷凝管、温度计、恒压滴液漏斗和磁力搅拌子的四口烧瓶中,在冰水浴和N2保护下搅拌均匀,并在2 h 内滴加丙烯酸甲酯(丙烯酸甲酯和EDA 的摩尔比为1∶8),升至30 ℃反应24 h,然后在50 ℃,133 Pa 下旋转减压蒸馏,除去过量的甲醇和丙烯酸甲酯,得到淡黄色黏稠状液体,即为半代超支化PAMAM。将半代超支化PAMAM和甲醇的混合溶液加入到四口烧瓶中,搅拌均匀并滴加EDA(EDA 和半代超支化PAMAM 的摩尔比为1∶16),在25 ℃下连续反应24 h,然后在72 ℃,266 Pa 下旋转减压蒸馏,除去过量的甲醇和EDA,得到黄色黏稠状液体,记为超支化PAMAM(EDA)。
按上述合成步骤,单体改用DETA,TETA可分别制备超支化PAMAM(DETA)和超支化PAMAM(TETA)。
将MAH 和去离子水加入到三口烧瓶中,充分溶解后滴加PAMAM(EDA)(PAMAM(EDA)和MAH 的摩尔比为10∶1),滴加完成后升至60 ℃,连续反应12 h,得到黏稠状深黄色油状液体,真空蒸发干燥2 h 除水,然后用丙酮多次洗涤,除去未反应的MAH,在恒温干燥箱中干燥24 h,得到产物记为端羧基超支化PAMAM(EDA)-COOH。按相同步骤可制备端羧基超支化PAMAM(DETA)-COOH 和端羧基超支化PAMAM(TETA)-COOH。
端羧基超支化PAMAM 合成原理见图1。从图1可看出,超支化PAMAM 具有3 种结构单元:树枝状支化(B)单元、末端(T)单元和线型(L)单元[10-11]。PAMAM(EDA)-COOH,PAMAM(DETA)-COOH,PAMAM(TETA)-COOH 的分子量分别为1 301,1 869,2 525。
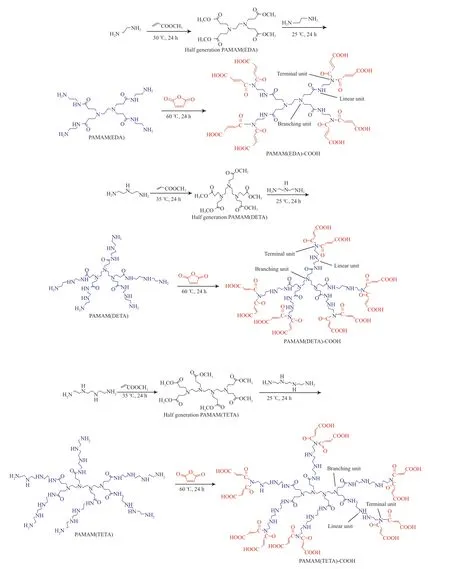
图1 超支化PAMAM 的合成路线Fig.1 Synthesis routes of hyperbranched polyamide-amines(PAMAM).
1.3 结构表征
采用FTIR 对试样结构进行表征,KBr 压片,波长500 ~4 000 cm-1,扫描次数32、分辨率4 cm-1;采用元素分析仪测定试样中元素的含量,分析试样纯度;采用固体1H NMR 测试试样的超支化结构;采用TG 分析试样的热稳定性。
1.4 阻垢性能评价
按SY/T 5673—2020[12]和GB/T 7476—1987[13]规定的方法测试阻垢性能。
1.5 分散性能测试
取FeSO4·7H2O 配制成10 mg/L Fe2+溶液,并用四硼酸钠溶液调节pH=11,然后分别向溶液中加入阻垢剂溶液和氯化钙溶液(Ca2+含量150 mg/L),搅拌均匀后,在恒温水浴中50 ℃下静置6 h,冷却至室温后,取少量上层清液于比色皿中,用紫外-可见分光光度计测量波长420 nm 处透过率,用Zeta 电位仪测试电位[14-15]。
1.6 钙垢晶体微观形貌SEM 分析
将CaSO4垢和CaCO3垢用去离子水冲洗多次后,在真空干燥箱中70 ℃下干燥24 h,然后观察钙垢的微观形貌。
2 结果与讨论
2.1 结构表征结果
2.1.1 FTIR 表征结果
不同端羧基超支化PAMAM 的FTIR 谱图见图2。由图2 可知,在3 350 ~3 310 cm-1处吸收峰较宽,归属于仲胺(N—H)的伸缩振动;2 940,2 840 cm-1处为亚甲基(—CH2)的对称伸缩振动和不对称伸缩振动吸收峰;1 710,1 410 cm-1处为羧基—C=O 键和C—H 键的伸缩振动峰;1 650,1 590 cm-1处出现了仲酰胺C=O 键的伸缩振动和N—H键的弯曲振动吸收峰;1 650 ~1 500 cm-1处的吸收峰还可能归属于C=C 双键;1 360 ~1 180 cm-1处的吸收峰归属于叔胺C—N 键的伸缩振动。FTIR表征结果显示,合成的3 种端羧基超支化PAMAM为目标产物。
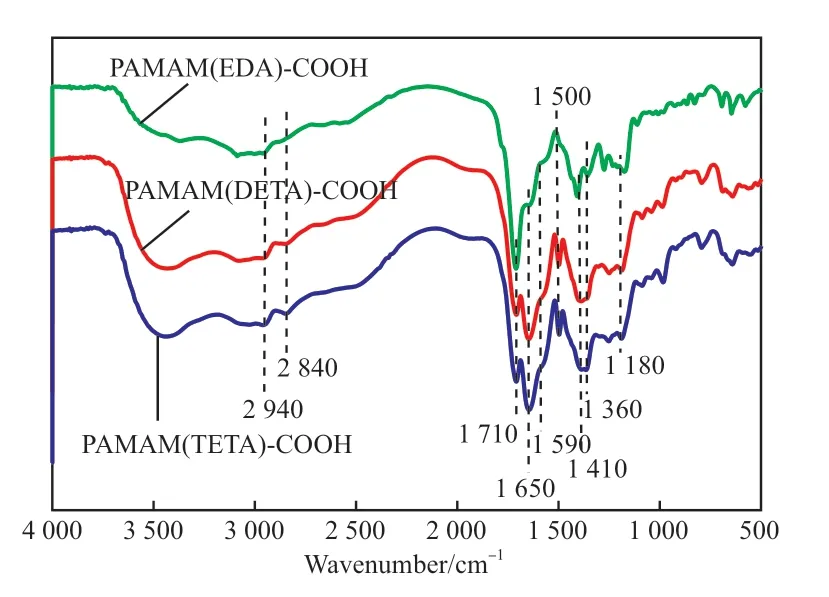
图2 不同端羧基超支化PAMAM 的FTIR 谱图Fig.2 FTIR spectra of different carboxyl-terminated hyperbranched PAMAM.
2.1.2 元素分析结果
不同端羧基超支化PAMAM 的元素分析结果见1。从表1 可看出,PAMAM(EDA)-COOH,PAMAM(DETA)-COOH,PAMAM(TETA)-COOH 中C 含量的测定值低于理论值,而H,N,O 含量的测定值高于理论值,产生该现象的原因为:1)在旋蒸、提纯过程中有EDA,DETA,TETA和马来酸等小分子未能除去;2)具有中心核并向外发散的超支化分子结构,在反应条件、空间位阻等影响下产生了桥接、环化等副反应。因此产物中元素含量的测定值与理论值不符,但差别并不大,仍可以确定为目标产物。
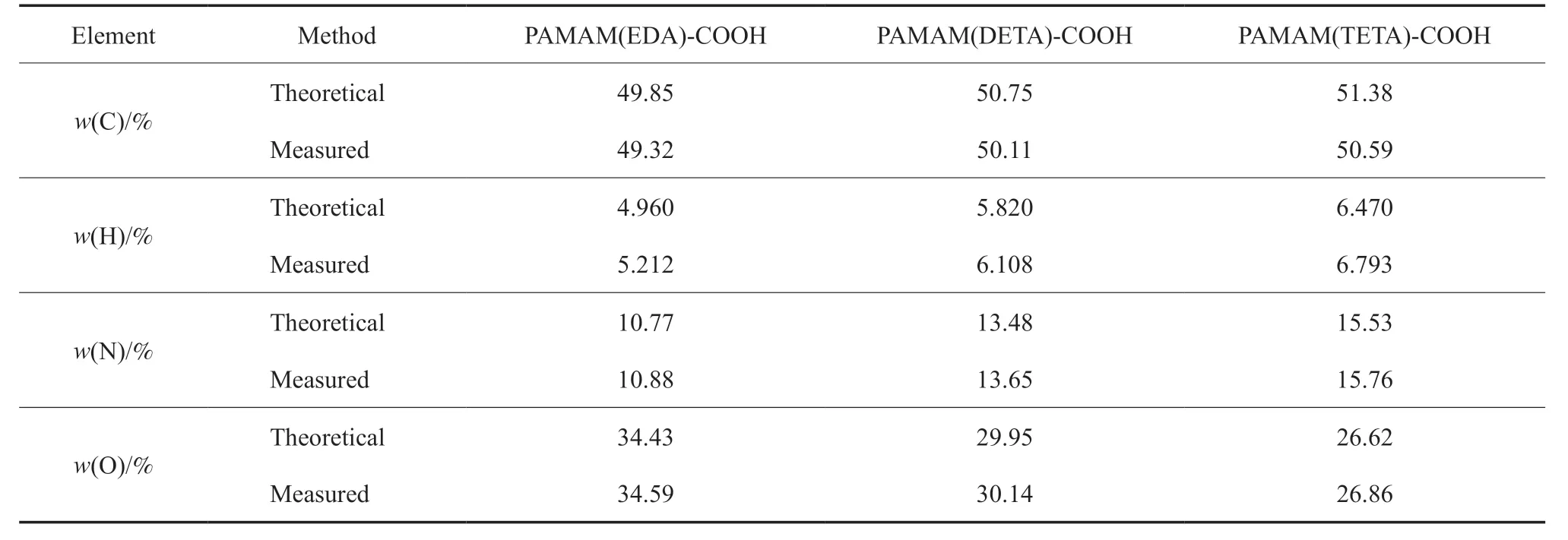
表1 不同端羧基超支化PAMAM 的元素分析结果Table 1 Elemental analysis of different carboxyl-terminated hyperbranched PAMAM
2.1.31H NMR 表征结果
不同端羧基超支化PAMAM 的1H NMR 谱图见图3。从图3 可看出,制备的3 种端羧基超支化PAMAM 具有相似的超支化结构,谱图中均含8 种峰,表明有8 种不同状态的H。化学位移δ为:3.31(a),6.63(b),6.14(c),2.80(d),2.92(e),3.12(f),2.59(g),2.50(h)。结合超支化结构的3 种重复单元可知,g,h 处的H 原子归属于B 单元;d,e,f 处的H 原子归属于L 单元;a,b,c处的H 原子归属于T 单元,其中a 处的H 原子归属于羧基。通过峰面积计算支化度,得到PAMAM(EDA)-COOH,PAMAM(DETA)-COOH,PAMAM(TETA)-COOH 的支化度分别为0.71,0.76,0.85,均高于普通超支化聚合物的支化度(0.50),这是因为末端结构单元上的H 被羧酸化后形成树枝状支化单元,从而导致制备的端羧基超支化PAMAM 的支化结构增多(即支化度增大)。
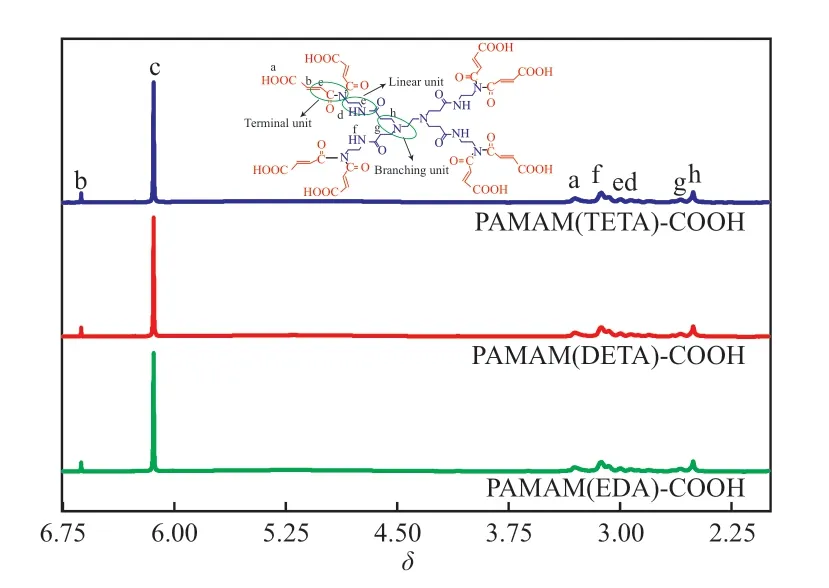
图3 端羧基超支化PAMAM 的1H NMR 谱图Fig.3 1H NMR spectra of carboxyl-terminated hyperbranched PAMAM.
2.1.4 TG 分析结果
不同端羧基超支化PAMAM 的TG 曲线见图4。由图4 可知,试样的TG 曲线均分为3 个阶段。PAMAM(EDA)-COOH 的第1 个阶段在0 ~209℃,质量损失15%;PAMAM(DETA)-COOH的第1 个阶段在0 ~309 ℃,质量损失20%;PAMAM(TETA)-COOH 的第1 个阶段在0 ~382℃,质量损失30%。可以看出支化结构增多,质量损失变大,这是因为在提纯过程中存在小分子,这些小分子包裹在支化分子内部,随着温度的升高先发生溶解,此阶段产物中还包含未完全蒸发的甲醇、水,以及接枝MAH 中的羧基分解和支化分子的断裂。PAMAM(EDA)-COOH 的第2 个阶段在209 ~414 ℃,质量损失25%;PAMAM(DETA)-COOH 的第2 个阶段在309 ~507 ℃,质量损失53%;PAMAM(TETA)-COOH 的第2个阶段在382 ~419 ℃,质量损失35%。可以看出在200 ℃以上,质量损失呈无规则性,而在此阶段主要是聚合物中—CONH—键、—C—C—键及—C=C—键的分解。PAMAM(EDA)-COOH 的第3 个阶段在414 ~610 ℃,质量损失41%;PAMAM(DETA)-COOH 的 第3 个 阶 段 在507 ~696 ℃,质量损失17%;PAMAM(TETA)-COOH 的第3 个阶段在419 ~560 ℃,质量损失35%;在此阶段主要是胺类的键断裂生成气体以及焦炭的分解。
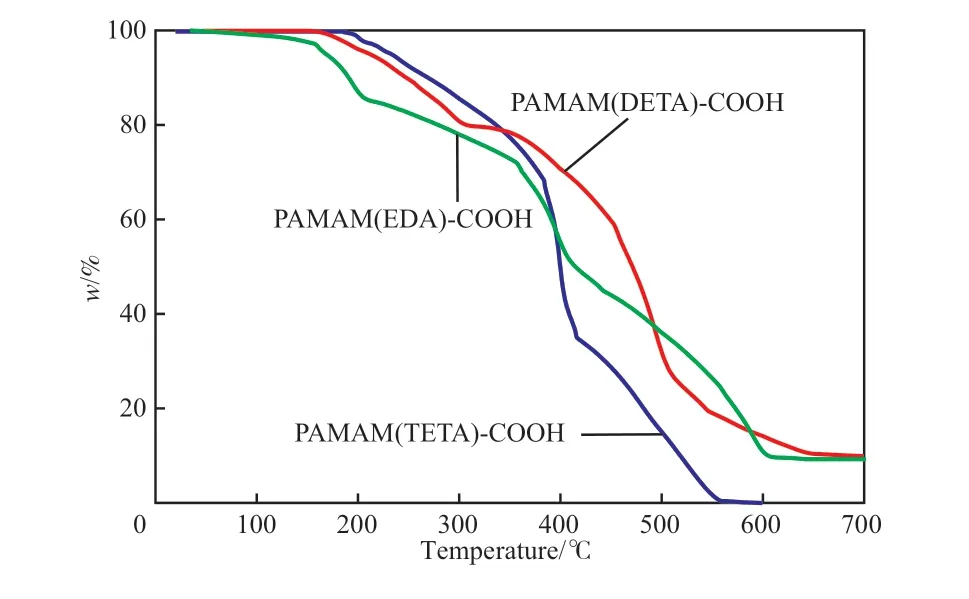
图4 不同端羧基超支化PAMAM 的TG 曲线Fig.4 TG curves of carboxyl-terminated hyperbranched PAMAM.
在静态钙垢的抑制实验中,测试温度一般在50 ~80 ℃[16]。而在50 ~100 ℃范围内3 种端羧基超支化PAMAM 的质量损失率均低于2%,说明它们具有良好的热稳定性。
2.2 阻垢性能测试
2.2.1 投加量对阻垢性能的影响
端羧基超支化PAMAM 阻垢剂在不同添加量下对钙垢的阻垢性能见图5。从图5 可看出,在投加量为100 ~400 mg/L 时,阻垢剂的阻垢率随投加量的增加而增大,在投加量为400 mg/L 时,PAMAM(EDA)-COOH,PAMAM(DETA)-COOH,PAMAM(TETA)-COOH 阻垢剂对CaSO4垢的阻垢率分别为96.4%,95.1%,94.9%,对CaCO3垢的阻垢率分别为90.2%,89.9%,88.9%,远高于SY/T 5673____2020[12]规 定 的CaSO4垢 或CaCO3垢的阻垢率。在投加量为400 ~800 mg/L 时,端羧基超支化PAMAM 阻垢剂对CaSO4垢的阻垢率下降,且随支化结构的增多,阻垢率下降趋势越明显,这可能是枝接的大量羧基产生了阈值效应[17],但此时对CaCO3垢的阻垢率达到了动态平衡。
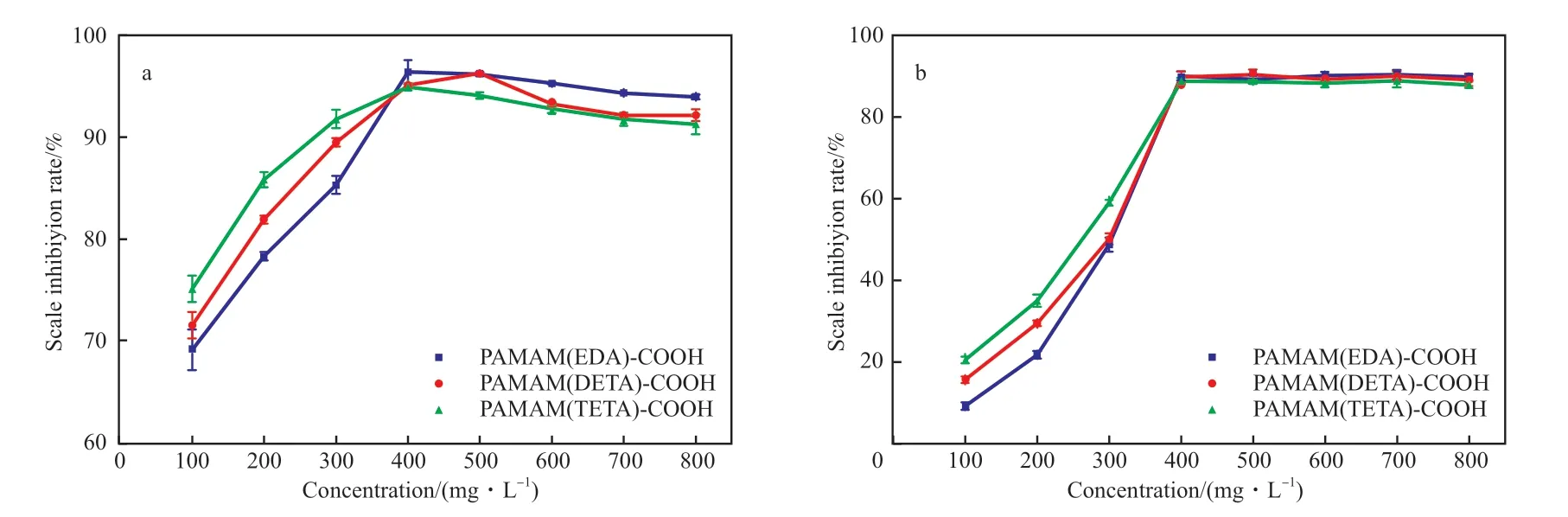
图5 阻垢剂阻垢性能随阻垢剂投加量的变化Fig.5 Variation of scale inhibition performance with scale inhibitor amount.
2.2.2 温度对阻垢性能的影响
温度对端羧基超支化PAMAM 阻垢剂阻垢性能的影响见图6。由图6 可知,随温度的升高,阻垢剂的阻垢率呈先增长后下降的趋势,在50 ℃时,PAMAM(EDA)-COOH,PAMAM(DETA)-COOH,PAMAM(TETA)-COOH 阻垢剂的阻垢率达到最高,对CaSO4垢的阻垢率分别为97.9%,96.4%,94.8%,对CaCO3垢的阻垢率分别为90.4%,89.2%,88.8 %。此后随着温度的继续升高,溶液中分子热运动加剧、相互碰撞速率加快[18],导致钙垢晶体提前析出,80 ℃时,PAMAM(EDA)-COOH,PAMAM(DETA)-COOH,PAMAM(TETA)-COOH阻垢剂对CaSO4垢的阻垢率分别为95.5%,93.8%,90.3%,对CaCO3垢的阻垢率分别为85.2%,84.1%,83.6%,可见高温下具有超支化结构的阻垢剂具有一定的耐高温性能,随支化结构的增多,阻垢率有所下降,但降幅不大,说明支化结构对高温下端羧基超支化PAMAM 阻垢剂的阻垢性能影响并不太大。
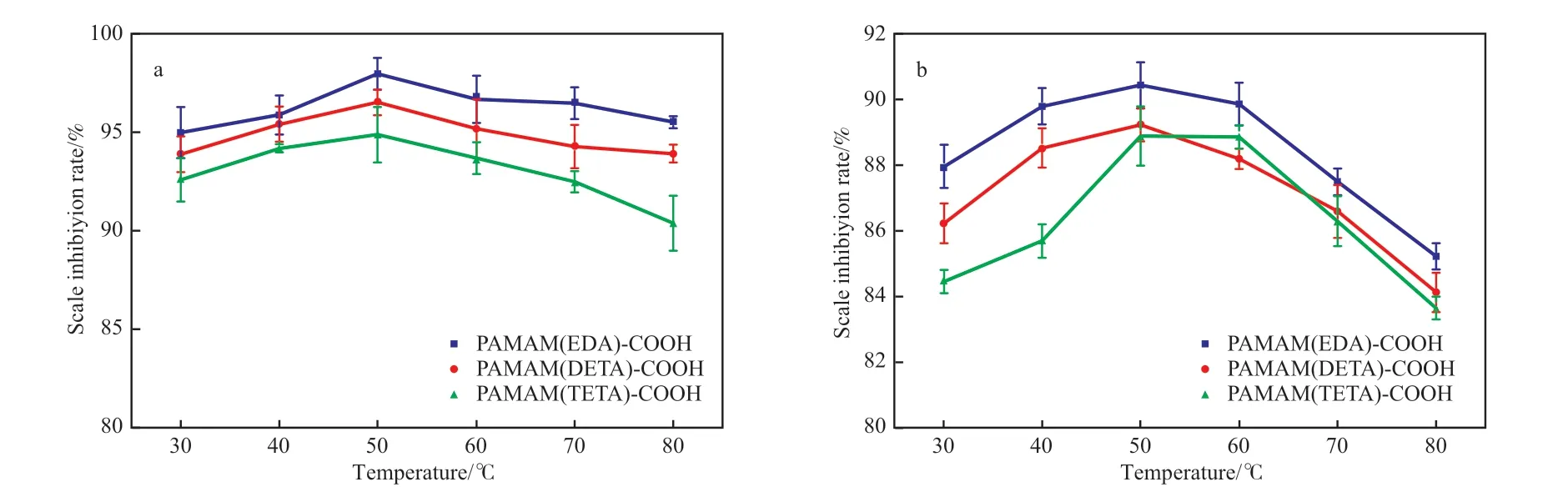
图6 温度对阻垢剂阻垢性能的影响Fig.6 Effects of temperature on scale inhibition performance of scale inhibitor.
2.2.3 溶液pH 对阻垢性能的影响
溶液pH 对端羧基超支化PAMAM 阻垢剂阻垢性能的影响见图7。由图7 可知,阻垢剂在酸性溶液中阻垢性能优异,但随pH 的增加阻垢率降低,这是因为在碱性状态下,盐溶液中的OH-先与Ca2+形成微溶性Ca(OH)2,Mg(OH)2沉淀,阻碍了超支化PAMAM 分子在CaSO4,CaCO3垢晶体表面的吸附。但随支化结构的增加,这有利于阻垢性能的提高,在溶液pH=10 时,3 种端羧基超支化PAMAM 阻垢剂对CaSO4垢的阻垢率均在80%以上,对CaSO4垢的阻垢率也在70 %以上。说明端羧基超支化PAMAM 具有较广的pH 应用范围。
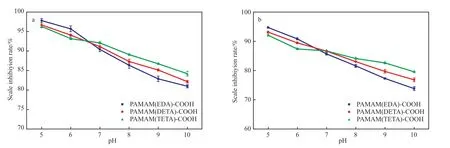
图7 溶液pH 对阻垢剂阻垢性能的影响Fig.7 Effects of solution pH on scale inhibition performance of scale inhibitor.
2.2.4 反应时间对阻垢性能的影响
反应时间对端羧基超支化PAMAM 阻垢剂阻垢性能的影响见图8。从图8 可看出,在0 ~6 h时,阻垢剂的阻垢率快速增加,在0 ~12 h 时,PAMAM(TETA)-COOH 阻垢剂的阻垢率增长最快,PAMAM(EDA)-COOH 阻垢剂的阻垢率增长最慢,并且随支化结构的增加,阻垢率增大,这是因为分子量越大,阻垢剂在溶液中的分散速率越快,越易于快速阻垢。在6 ~18 h 时,阻垢率缓慢增加,在18 ~36 h 时,阻垢率基本保持不变,这说明具有超支化结构的聚合物阻垢性能稳定。在36 h 时,3 种端羧基超支化PAMAM 阻垢剂对CaSO4垢的阻垢率均大于94%,对CaSO4垢的阻垢率均大于88%。
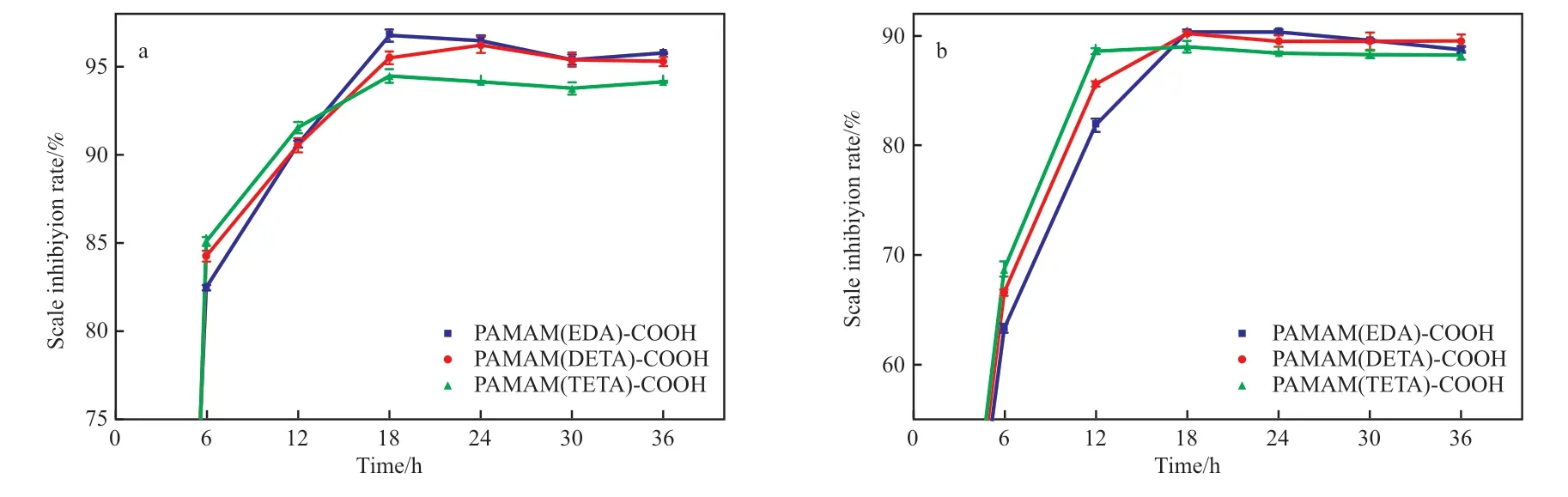
图8 反应时间对阻垢剂阻垢性能的影响Fig.8 Effects of reaction time on scale inhibition performance of scale inhibitor.
2.2.5 与其他类型阻垢剂对比
将所制备的阻垢剂与市售阻垢剂聚环氧琥珀酸(PESA),羟基乙叉二膦酸(HEDP),MAH进行对比,结果见图9。在图9 可看出,制备的3种端羧基超支化PAMAM 阻垢剂对CaSO4,CaCO3垢的阻垢率均高于PESA,HEDP,MAH。MAH在投加量为600 mg/L 时,未检测出对钙垢的阻垢率,这可能是配制的标准盐水溶液中矿化度较高,影响了MAH 在溶液中的分散,同时也说明端羧基超支化PAMAM 阻垢剂可应用于高矿化度水的复杂环境中。
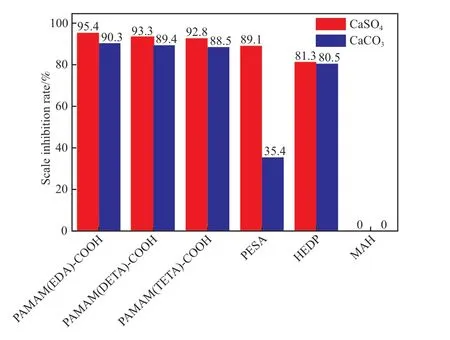
图9 不同阻垢剂的阻垢率Fig.9 Scale inhibition rate of different scale inhibitors.
2.3 分散性能测试
端羧基超支化PAMAM 的分散性能见表2。从表2 可看出,同一浓度下透过率的大小顺序为:PAMAM(EDA)-COOH>PAMAM(DETA)-COOH>PAMAM(TETA)-COOH,均低于MAH的透过率;而Zeta 电位绝对值的大小顺序为:
PAMAM(EDA)-COOH 钙垢晶体的SEM 照片见图10 ~11。由图10可知,CaSO4垢晶体呈长方形状、表面光滑和结构完整,加入端羧基超支化PAMAM 阻垢剂后,CaSO4垢晶体表面变得非常粗糙,出现不同程度裂痕并且有小晶体从表面脱落,CaSO4垢晶体结构遭到严重破坏,力学性能降低,形成的钙垢晶体不易附着在设备表面。由图11 可知,CaCO3垢晶体表面光滑、具有规则外形和致密结构,且这种晶型的晶体易于团聚。在加入端羧基超支化PAMAM 阻垢剂后,CaCO3垢晶体的晶型发生转变,生长出现紊乱状态,晶体表面凹凸不平,密度降低并且开始分散。产生该现象的原因是超支化结构聚合物具有更大的有效比表面积,占据了CaCO3垢晶体的活性增长位点,从而导致晶格畸变。另外端羧基超支化PAMAM 的分子外围有大量羧基,它们可与溶液中更多的钙离子螯合,进一步阻碍晶体的正常生长顺序。 图10 CaSO4 垢晶体的SEM 照片Fig.10 SEM images of CaSO4 scale crystal. 图11 CaCO3 垢晶体的SEM 照片Fig.11 SEM images of CaCO3 scale crystal. 通过对制备的端羧基超支化PAMAM 的阻垢性能、分散性能和垢样的分析,发现阻垢机理主要与超支化分子结构、末端基官能团有关,见图12。从图12 可看出,由于钙垢晶体的形成主要是溶液中存在大量Ca2+,Mg2+,HCO3-,CO32-,SO42-等离子,这些离子在溶液中发生碰撞形成晶核小分子,而超支化聚合物具有三维网状结构,向外发散形成的纳米级内表面可包裹和吸附小分子,抑制或降低离子碰撞的概率,从而减少晶体成核;在碱性环境下,支化度越高的端羧基超支化PAMAM 的分散阻垢性能越强,这是因为支化单元链上存在大量仲胺。改性超支化PAMAM 接枝大量的羧基,分子量变大、分散性能增强,从而可以更好地吸附和分散微晶,而且超支化PAMAM 外层带有大量负电荷,从而形成双电子层结构,在静电作用下与同类型离子发生排斥,从而不能使钙垢晶体正常生长。 图12 阻垢机理示意图Fig.12 Schematic diagram of scale inhibition mechanism. 1)利用Michael 加成和酰胺化反应合成了多乙烯多胺类超支化聚合物,再用MAH 封端剂改性,制备了端羧基超支化PAMAM 阻垢剂PAMAM·(EDA)-COOH,PAMAM(DETA)-COOH,PAMAM(TETA)-COOH,它们均具有良好的热稳定性。 2)在70 ℃、pH=7、16 h、投加量400 mg/L 的条件下,PAMAM(EDA)-COOH,PAMAM(DETA)-COOH,PAMAM(TETA)-COOH 阻垢剂对CaSO4垢的阻垢率达到96.4%,95.1%,94.9%,对CaCO3垢的阻垢率达到90.2%,89.9%,88.9%,优于市售阻垢剂PESA,HEDP,MAH。同时,合成的端羧基超支化PAMAM 阻垢剂耐高温、pH 应用范围广、分散性能优异。 3)加入端羧基超支化PAMAM 阻垢剂后使钙垢晶体受严重破坏,出现晶型转变和晶格畸变现象,从而在一定程度上抑制了钙垢晶体生长。推测阻垢机理主要与超支化分子结构和末端基团有关。2.4 垢样的SEM 表征结果


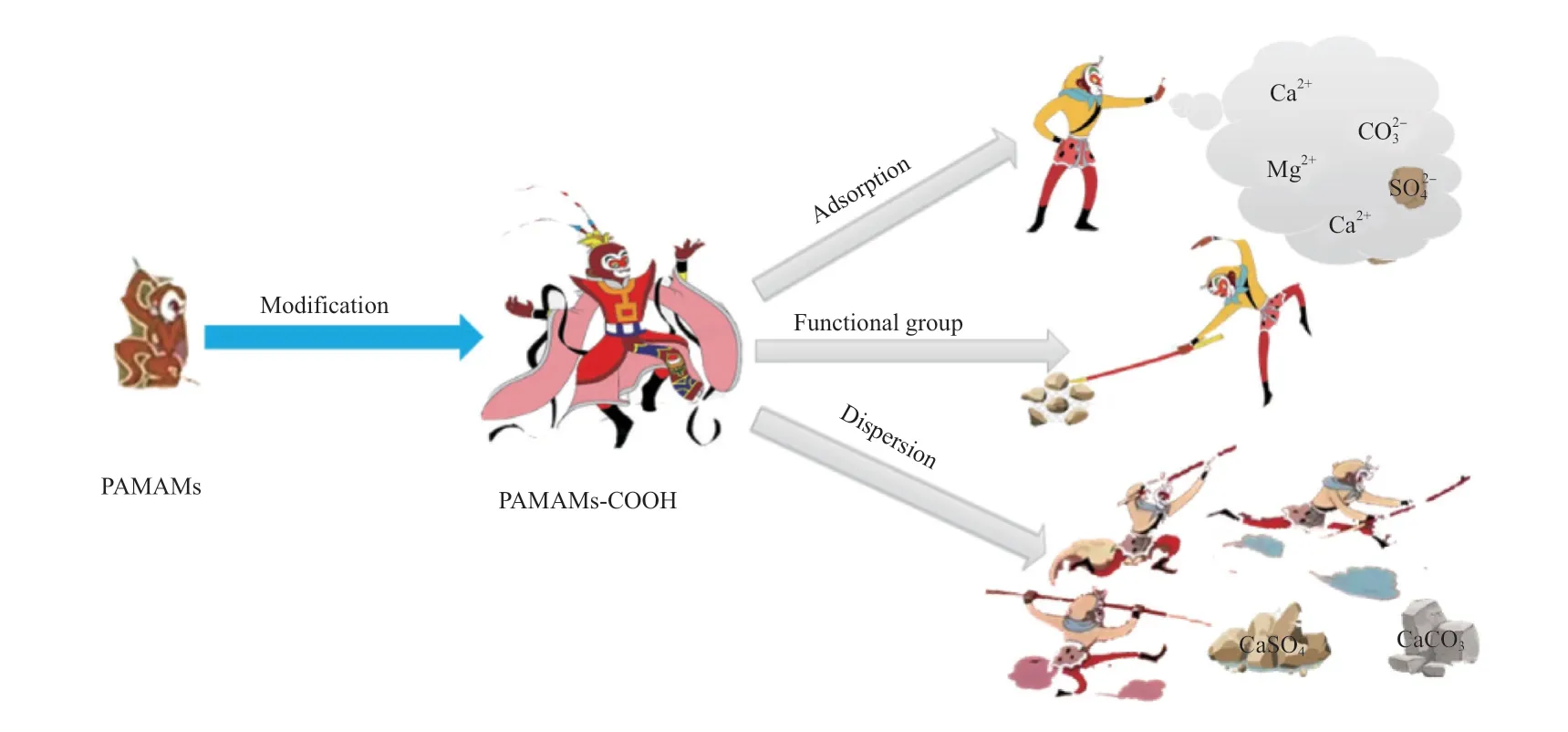
2.5 阻垢机理推测
3 结论
猜你喜欢
工业安全与环保(2022年10期)2022-10-28
中原工学院学报(2022年3期)2022-09-01
浙江大学学报(理学版)(2020年1期)2020-03-12
天津城建大学学报(2015年5期)2015-12-09
安徽农学通报(2015年2期)2015-02-12
化工管理(2014年24期)2014-12-11
应用化工(2014年10期)2014-08-16
应用化工(2014年9期)2014-08-10
应用化工(2014年7期)2014-08-09
制冷学报(2014年1期)2014-03-01
