
枸杞糖肽通过抑制铁死亡减轻高血糖大鼠脑缺血/再灌注损伤
2023-11-10 14:14马艳梅张建忠
中国药理学通报 2023年11期
李 鹏,杨 晶,马艳梅,张建忠,景 丽
(宁夏医科大学 1. 基础医学院病理学系、2. 总医院放射科,宁夏 银川 750004)
高血糖是急性缺血性卒中(acute ischemic stroke,AIS)的独立危险因素[1]。与正常血糖水平患者相比,高血糖对卒中患者的影响体现在疾病诊疗的全过程,包括更严重的临床表现、更广泛的梗死面积和更高的病死率、致残率及复发率[2]。研究高血糖状态下脑缺血/再灌注损伤(cerebral ischemia/reperfusion injury,CIRI)的发病机制及其有效的防治策略对于合并高血糖AIS患者的综合管理具有重要意义。近年来,越来越多的研究发现,铁死亡在CIRI中发挥重要作用,抑制铁死亡可能减轻CIRI[3-4]。
枸杞具有抗氧化应激、抗炎、抗肿瘤、抗衰老、免疫调节,以及保护心脑血管疾病等作用[5]。枸杞糖肽(Lyciumbarbarumglycopeptide,LbGp)是从枸杞多糖中进一步分离出的一种具有免疫活性的糖缀合物,包含5种组分,其分子量为88 ku。研究显示,枸杞多糖通过提高超氧化物歧化酶(superoxide dismutase,SOD)和谷胱甘肽过氧化物酶的活力,降低丙二醛(malondialdehyde,MDA)含量,进而抑制氧化应激发挥CIRI后的脑保护作用[6]。由于铁死亡是由氧化应激诱导的一种独特的程序性细胞死亡形式,我们推测LbGp可能具有调控铁死亡的作用。然而,LbGp是否能够通过抑制铁死亡减轻高血糖状态下的CIRI,目前尚不清楚。因此,本研究采用线栓法建立体内慢性高血糖大鼠左侧短暂性大脑中动脉栓塞再灌注(transient middle cerebral artery occlusion/reperfusion,tMCAO/R)模型,模拟临床高血糖状态下AIS患者机械取栓手术后的脑缺血/再灌注病理生理过程,并使用铁死亡抑制剂去铁酮(deferiprone,DFP)作为阳性对照药物,探讨LbGp是否通过调控铁死亡的相关靶点抑制铁死亡,减轻高血糖大鼠CIRI,以期为高血糖状态下CIRI的诊治提供新的思路和方向。
1 材料与方法
1.1 实验动物SD大鼠,♂,体质量(240~270) g,购自宁夏医科大学实验动物中心[生产许可证号:SCXK(宁)2020-0001]。大鼠饲养于实验动物中心SPF级动物房,室温(21~23) ℃,12 h/12 h光/暗交替,自由获取水和食物。本研究所有实验操作均经宁夏医科大学实验动物中心实验动物福利伦理委员会批准(编号:IACUC-NYLAC-2021-047)。
1.2 主要试剂与仪器LbGp粉末(中宁枸杞院士工作站宁夏天仁枸杞生物科技股份有限公司);去铁酮片(Apotex Inc., Etobicoke site);组织铁测定试剂盒(货号:A039-2-1),购自南京建成生物工程研究所;还原型谷胱甘肽(reduced glutathione,GSH)和氧化型谷胱甘肽(oxidized glutathione disulfide,GSSG)检测试剂盒、总SOD活性检测试剂盒、脂质氧化(MDA)检测试剂盒、总抗氧化能力(total antioxidant capacity,T-AOC)检测试剂盒(货号分别为S0053、S0101S、S0131S、S0121),均购自上海碧云天生物技术有限公司;转铁蛋白受体1(transferrin receptor 1,TFR1)抗体(货号:TA5343),购自Abmart;二价金属离子转运体1(divalent metal transporter 1,DMT1)抗体、膜铁转运蛋白1(ferroportin 1,FPN1)抗体(货号分别为20507-1-AP、26601-1-AP),均购自Proteintech公司;脂酰辅酶A合成酶长链家族成员4(acyl-CoA synthetase long-chain family member 4,ACSL4)抗体、二抗(货号分别为ab155282、ab6721),均购自Abcam公司;谷胱甘肽过氧化物酶4(glutathione peroxidase 4,GPX4)抗体(货号:NBP2-75511),购自Novus;β-actin抗体(货号:#4970S),购自Cell Signaling Technology公司。大鼠专用线栓(北京西浓科技有限公司);Amersham Imager 600化学发光成像系统、Signa Architet 3.0T MRI扫描仪(美国通用电气公司);石蜡切片机(德国Leica公司);光学及荧光显微镜(日本Olympus公司)。
1.3 大鼠高血糖模型的建立各组大鼠置于笼盒中预适应3 d,禁食12 h,称体质量,以60 mg·kg-1腹腔注射冰上现配的含链脲佐菌素(streptozotocin,STZ)的柠檬酸缓冲液,之后放回笼盒,自由进食和饮水。分别于3 d、7 d后,取大鼠尾静脉血,检测血糖值,以血糖值 >16.8 mmol·L-1为建立高血糖模型成功。
1.4 大鼠tMCAO/R模型的建立建立经左侧颈外动脉(external carotid artery,ECA)—颈内动脉(internal carotid artery,ICA)—大脑中动脉(middle cerebral artery,MCA)途径的tMCAO/R模型。首先,腹腔注射1%戊巴比妥钠(40 mg·kg-1)麻醉大鼠,麻醉后大鼠仰卧位固定;然后,颈部正中线略偏左侧行手术切口,依次钝性分离并暴露左侧颈总动脉(common carotid artery,CCA)、ICA和ECA,用小型动脉夹暂时夹闭CCA远端和ICA近端,在ECA两结之间用眼科剪剪一小口,并将适合体质量的线栓缓慢插入ECA,后经ICA入颅至MCA开口处,固定线栓;最后,在缺血30 min后将线栓轻轻拔出,确保拔出的线栓头部完整,结扎ECA,消毒并逐层缝合皮肤。假手术组除不插栓线外,其余步骤同tMCAO/R模型。
1.5 动物分组与给药大鼠随机分为高血糖假手术组(Sham,n=6)、高血糖tMCAO/R组(HG,n=8)、高血糖枸杞糖肽预处理tMCAO/R组(LbGp,n=8)、高血糖去铁酮阳性药物对照tMCAO/R组(DFP,n=8)。每组各有3只大鼠用于Western blot实验。本研究LbGp和DFP的给药时间点选择在注射STZ后的第8天,以更真实地模拟临床实际,大鼠每天灌胃1次,连续灌胃3周。每次灌胃前,将LbGp和DFP充分溶于大鼠无菌饮用水中。参照文献及预实验,LbGp的给药剂量为20 mg·kg-1·d-1[7],DFP的给药剂量为75 mg·kg-1·d-1[8]。
1.6 神经功能缺损评分采用改良的神经功能缺损评分(modified neurological severity score,mNSS)法[9],全面评价大鼠的神经功能缺损情况。mNSS项目主要包括运动实验(提尾实验和行走实验)、感觉实验(本体感觉实验和放置实验)、平衡木实验和反射实验4个方面。mNSS总分18分,0分为神经功能正常,18分为神经功能缺损最严重。tMCAO/R模型成功的判定标准为mNSS≥4分,且同时具备4个方面评分项目神经功能均有缺损。
1.7 脑组织铁含量、GSH/GSSG比值、SOD活性、T-AOC及MDA含量的检测均使用相应试剂盒检测,各实验步骤均严格按照说明书操作。
1.8 脑梗死体积测量采用磁共振T2加权成像(T2-weighted imaging,T2WI)法和TTC染色法测定脑梗死体积,通过ImageJ软件计算脑梗死体积百分比。T2WI检查:大鼠麻醉后,俯卧位固定于大鼠专用头部线圈内,行T2WI冠状位扫描,扫描范围包括嗅球至上段颈髓;扫描参数重复时间2 257 ms,回波时间84.6 ms,层厚2 mm,层数12,视野80 mm×80 mm,矩阵288×192;脑梗死区表现为异常高信号,正常脑实质表现为等信号。TTC染色:MRI检查后麻醉处死,迅速取脑,去除嗅球和小脑,-20 ℃冷冻20 min,行冠状面薄层切片后,放于新鲜配制的TTC溶液中,避光37 ℃孵育30 min,期间翻动2次,然后4%多聚甲醛溶液固定,过夜后拍照;TTC染色脑梗死区表现为白色,非梗死区表现为红色。
1.9 HE染色大鼠取材后,经固定、脱水、透明、浸蜡、包埋后制作成石蜡切片(厚约4 μm)。然后,依次经烘烤、脱蜡和水化,苏木精染色、盐酸乙醇分化、伊红染色,最后脱水、透明、封片,并于光学显微镜下观察脑组织病理学形态改变。
1.10 普鲁士蓝染色(增强型)切片依次经脱蜡、水化、Perls染色工作液、孵育工作液、增强工作液、复染、脱水、透明、封片后,于光学显微镜下观察。
1.11 免疫组织化学(immunohistochemistry,IHC)染色切片依次经脱蜡、水化、高压抗原修复、阻断内源性过氧化物酶和山羊血清封闭后,分别加入一抗TFR1、DMT1、FPN1(均1 ∶200),然后孵育二抗,最后显色、复染、脱水、透明和封片。置于400倍光学显微镜下观察,采用ImageJ软件测量目的蛋白的平均光密度值。
1.12 Western blot检测提取大鼠大脑缺血侧皮质总蛋白,并定量、变性。以10 μL体系上样,依次经电泳、转膜、封闭后,分别加入一抗TFR1(1 ∶1 000)、DMT1(1 ∶800)、FPN1(1 ∶1 000)、ACSL4(1 ∶10 000)、GPX4(1 ∶1 000)、β-actin(1 ∶1 000),然后二抗(1 ∶5 000)孵育、曝光。采用ImageJ软件分析目的蛋白的相对表达量。

2 结果
2.1 LbGp对高血糖tMCAO/R大鼠神经功能缺损和氧化应激损伤的影响大鼠脑缺血30 min,再灌注24 h后,Tab 1检测结果表明,Sham组大鼠mNSS为0,HG组大鼠mNSS明显升高(P<0.01);与HG组相比,LbGp组和DFP组大鼠mNSS均明显降低(P<0.01)。此外,与Sham组相比,HG组大鼠GSH/GSSG比值、SOD活性和T-AOC均明显降低(P<0.01),MDA含量明显升高(P<0.01);与HG组相比,LbGp组和DFP组大鼠GSH/GSSG比值、SOD活性和T-AOC均明显升高(P<0.05,P<0.01),MDA含量明显降低(P<0.01)。
2.2 LbGp对高血糖tMCAO/R大鼠脑梗死体积的影响基于动物福利,最大化减少动物实验数量。实验前先选择3只正常血糖大鼠,通过磁共振T2WI法和病理TTC染色法,观察脑梗死范围。结果显示(Fig 1),T2WI法和TTC染色法测量的脑梗死体积差异无统计学意义(P>0.05),因此后续实验选择磁共振T2WI法测量脑梗死体积。如Fig 2所示,Sham组大鼠T2WI左右两侧大脑半球脑实质信号正常,未见异常高信号,且中线结构居中,两侧侧脑室对称、无扩张表现;与Sham组相比,HG组、LbGp组和DFP组大鼠T2WI左侧大脑半球脑实质均出现不同程度的异常高信号,并以HG组信号强度最高(提示血管源性脑水肿最严重),且HG组伴有左侧脑室受压变窄、右侧脑室扩张积水及中线结构局部右偏。与HG组相比,LbGp组和DFP组脑梗死体积百分比均减小(P<0.01,P<0.05)。
2.3 LbGp对高血糖tMCAO/R大鼠脑组织病理学变化的影响如Fig 3所示,Sham组HE染色神经元核呈圆形,核仁居中,胞质丰富,胞质着色均匀;HG组神经元胞质染色变浅,大部分神经元呈空泡样改变,间质水肿明显;LbGp组和DFP组较HG组神经元形态改变和间质水肿程度有所缓解。与Sham组相比,HG组皮质缺血半暗带区核固缩神经元数量明显增多(P<0.01);与HG组相比,LbGp组和DFP组核固缩神经元数量均明显减少(P<0.01,P<0.05)。

Tab 1 Effects of LbGp on mNSS, GSH/GSSG ratio, SOD activity, T-AOC and MDA contents in hyperglycemic rats

Fig 1 Comparison of T2WI and TTC staining for showing cerebral infarction volume in normoglycemic rats ( n=3)

Fig 2 Effect of LbGp on cerebral infarction volume in hyperglycemic rats n=3~5)
2.4 LbGp对高血糖tMCAO/R大鼠脑组织铁含量及铁代谢相关蛋白的影响普鲁士蓝染色结果(Fig 4A)显示,Sham组大鼠脑皮质区可见散在分布的点片状棕褐色染色,HG组大鼠缺血皮质区可见多发点片状或小片状棕褐色染色,LbGp组和DFP组缺血皮质区均未见异常的棕褐色染色。各组大鼠脑组织铁含量测定结果(Fig 4B)显示,与Sham组相比,HG组脑组织铁含量明显增加(P<0.01);与HG组相比,LbGp组和DFP组脑组织铁含量均明显降低(P<0.01)。
IHC结果显示(Fig 5),与Sham组相比,HG组TFR1、DMT1表达量均明显升高(P<0.05,P<0.01),FPN1表达量明显下降(P<0.01);与HG组相比,LbGp组和DFP组TFR1、DMT1表达量均明显下降(P<0.01,P<0.05),FPN1表达量均明显升高(P<0.01)。Western blot结果显示(Fig 6),各组TFR1、DMT1和FPN1蛋白的表达趋势与IHC结果基本一致。
2.5 LbGp对高血糖tMCAO/R大鼠ACSL4和GPX4蛋白表达的影响Western blot检测结果显示(Fig 7),与Sham组相比,HG组大鼠ACSL4蛋白表达明显升高,GPX4蛋白表达明显降低(P<0.01);与HG组相比,LbGp组和DFP组ACSL4蛋白表达明显降低,GPX4蛋白表达明显升高(P<0.05,P<0.01)。
3 讨论
本研究采用从ECA插线栓的路径,能够最大化比拟临床机械取栓手术。结果表明,高血糖大鼠脑缺血30 min再灌注24 h出现了占位性脑水肿征象,而经LbGp干预后,脑水肿程度明显减轻。组织病理学结果也证实,LbGp能够通过减少核固缩神经元数量、减轻细胞及间质水肿,改善高血糖大鼠脑缺血/再灌注诱导的神经元损伤。
氧化应激是导致AIS神经元功能紊乱和死亡的主要诱发因素。铁死亡作为一种由氧化应激诱导的独特程序性细胞死亡,其特点是脂质过氧化和GSH耗竭[10]。本研究结果显示,LbGp通过提高GSH/GSSG比值、SOD活性和T-AOC,减轻高血糖脑缺血/再灌注诱导的氧化应激损伤。正常情况下,细胞外游离的Fe3+与转铁蛋白(transferrin,TF)结合形成复合物,被细胞膜上的TFR1识别,并进一步形成TF-TFR1复合物。TF-TFR1内吞入细胞后,Fe3+从TF中释放出来,被前列腺跨膜上皮3抗原抗体还原为Fe2+。随后,Fe2+通过DMT1运输到细胞质中的不稳定铁池[11]。细胞内的Fe2+可通过细胞膜上的FPN1外排输出细胞,FPN1是目前唯一已知的铁外排蛋白[11]。细胞内过量的Fe2+可以催化H2O2生成羟自由基,并可通过芬顿反应和铁依赖性酶产生大量的活性氧,造成细胞膜的氧化损伤[12]。此外,铁是脂氧合酶催化亚基的重要组成部分,脂氧合酶活性的增加可介导脂质过氧化[13]。因此,过量的铁可诱导铁死亡的发生。本研究结果提示,高血糖脑缺血/再灌注大鼠脑组织出现了铁超载和铁代谢紊乱,而LbGp能够改善高血糖脑缺血/再灌注诱导的铁超载和铁代谢紊乱。为了进一步验证铁死亡是否参与了高血糖状态下脑缺血/再灌注诱导的脑损伤,本研究检测了铁死亡的其他重要调控因子——ACSL4和GPX4。ACSL4被认为是决定铁死亡敏感性的一个关键因子,其可调节多不饱和脂肪酸的合成,催化花生四烯酸或肾上腺酸酯化为磷脂酰乙醇胺[14],再经过一系列的生化反应,氧化为亲铁性的脂质过氧化物而激活细胞发生铁死亡[15]。研究显示,敲除ACSL4可以减轻小鼠CIRI,而过表达ACSL4则会通过诱导铁死亡加重CIRI[16]。GPX4作为一种含硒蛋白,在调控铁死亡方面起着关键作用,它能将有毒的膜脂H2O2还原成无毒的脂质醇[17]。Hu等[18]研究显示,β-石竹烯通过下调ACSL4的表达、上调GPX4的表达,抑制铁死亡,从而发挥对大鼠CIRI的脑保护作用。本研究结果显示,LbGp能够通过抑制铁死亡,减轻高血糖大鼠CIRI。
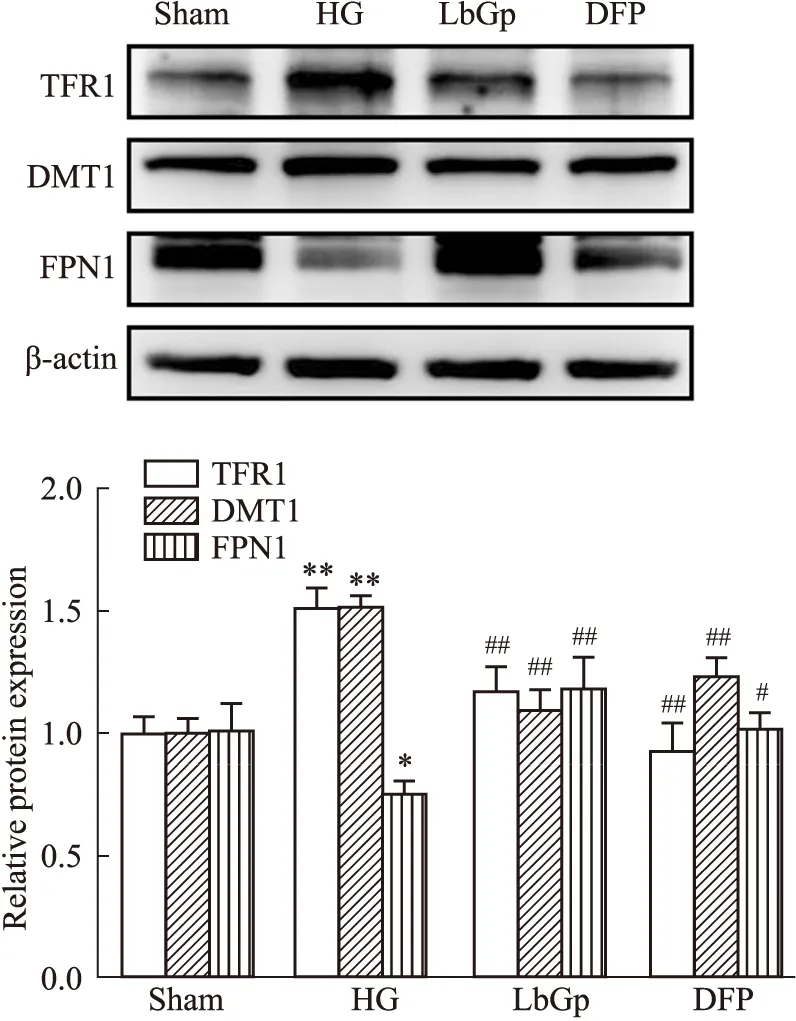
Fig 6 Effects of LbGp on TFR1, DMTI and FPN1 protein expressions in hyperglycemic rats n=3)
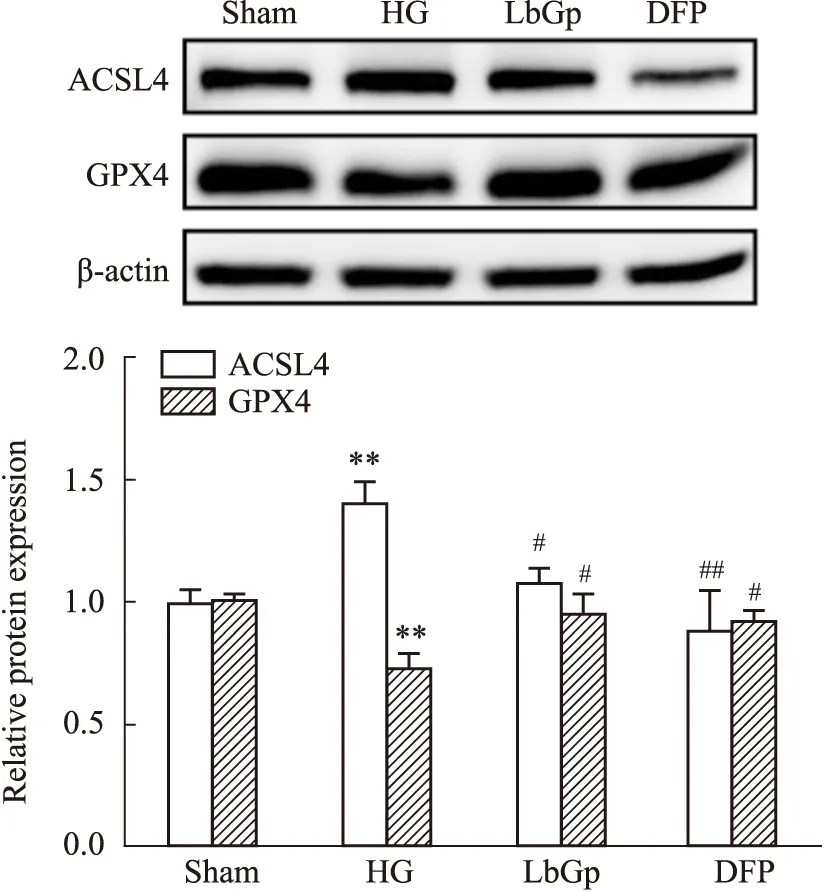
Fig 7 Effects of LbGp on protein expressions of ACSL4 and GPX4 in hyperglycemic rats n=3)
综上所述,本研究通过建立体内CIRI动物模型,证实LbGp能够减轻高血糖大鼠CIRI。之后通过检测氧化应激、铁含量和铁代谢相关蛋白,以及ACSL4、GPX4蛋白表达等多种铁死亡关键指标,证实LbGp可通过调节氧化应激、铁稳态和铁死亡相关蛋白的表达,抑制高血糖大鼠脑缺血/再灌注诱导的铁死亡。但本研究未能进一步探讨LbGp抑制铁死亡,减轻高血糖大鼠CIRI的具体作用机制,今后应从体内和体外两个实验水平进行深入研究。
猜你喜欢
自我保健(2021年4期)2021-06-16
世界科学技术-中医药现代化(2020年2期)2020-07-25
西南军医(2016年6期)2016-01-23
中国卫生标准管理(2015年15期)2016-01-15
中国医学装备(2015年10期)2015-12-29
中国康复理论与实践(2015年10期)2015-12-24
吉林大学学报(医学版)(2015年5期)2015-12-16
中国体外循环杂志(2015年3期)2015-12-08
中国当代医药(2015年23期)2015-03-01
西南军医(2015年2期)2015-01-22
