
雌性激素受体甲基化与中枢神经系统疾病的研究进展
2023-11-10 13:31高冰清代巧妹
中国药理学通报 2023年11期
刘 宏,黄 杨,温 薇,赵 莉,杨 婧,高冰清,代巧妹
(黑龙江中医药大学 1. 基础医学院、2. 研究生学院,黑龙江 哈尔滨 150040; 3大连医科大学中西医结合学院, 辽宁 大连 116044)
雌激素是一种重要的生理激素,在体内各生理系统均发挥着重要的调节作用,雌激素主要通过雌激素受体蛋白所介导的雌激素受体发挥生理功能[1]。雌激素受体在体内各处均有表达,同时也可以调节大脑结构和功能,增加神经元数量,改变树突结构,修复突触受损部位,发挥神经保护作用。近年来,表观遗传领域颇受青睐,在各种疾病发展过程中也常可寻其身影。随着雌激素受体甲基化研究日益深入,人们对其在中枢神经系统中的作用愈加关注,虽然甲基化检测手段日新月异,但雌激素受体甲基化具体作用却不甚明确。本文就雌激素受体的结构、分布、甲基化过程以及在不同中枢神经系统疾病过程中发挥的作用进行总结与阐述。
1 雌激素及其受体甲基化
1.1 雌激素及其受体雌激素是一种甾体激素,属于类固醇,其合成主要位于卵泡内,少部分在脑中进行。目前发现的雌激素受体主要包括核受体和膜受体。
1.1.1经典的核受体(ERα和ERβ) 典型的雌激素核受体(ERα和ERβ)包含了A、B、C、D、E、F 5种功能区域,但两者结构不全然相同。A/B区位于氨基末端结构域(n-terminal domain, NTD),是转录激活功能区(AF-1),同时也是两种受体结构差异最大的区域。C区对应于DNA结合结构域(DNA-binding domain, DBD),在序列特异性DNA结合和受体二聚化中具有重要作用,ERα和ERβ在此有97%的同源性。D结构域是连接C和E结构域的铰链区域,作用是和DNA结合。羧基末端是E / F区域,也称为配体结合域,即雌激素应答元件。包含雌激素结合区,以及共激活剂和核心加压剂的结合位点[2]。最后,雌激素受体转录活性的另外两个调节因子称为激活功能(AF)结构域AF1和AF2,分别位于NTD和配体结合域(ligand-binding domain,LBD)内。两受体功能区域和结构部位氨基酸(amino acid,AA)长度对比。
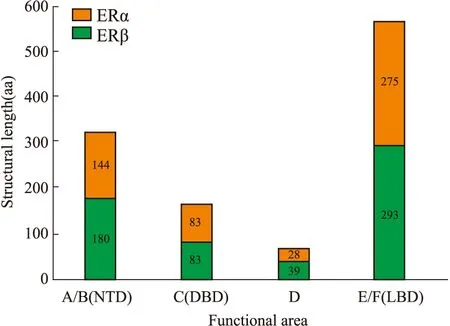
Fig 1 Structural diagram of various parts of estrogen nuclear receptor
1.1.2膜性受体 膜性受体含有经典核受体的膜性成分以及属于G蛋白偶联受体家族的GPER1(GPR30)、Gaq-ER和ER-X。其中GPR30发挥作用最为突出,GPR30的基因位于7号染色体(位点7p22.3)。就结构而言,GPER1与ERα或ERβ没有相似之处。作为典型的G蛋白偶联受体,其结构由7个跨膜α-螺旋区、4个细胞外段和4个胞质段组成,其DNA 序列的长度为2 604 bp,其中包含一个开放阅读框(其长度1 128 bp)。与其他雌激素受体相比,该受体对雌二醇(E2)具有较低的结合亲和力,但分布较更为广泛。除雌激素及其作用的靶器官和细胞膜、内质网、线粒体及高尔基复合体外,在骨骼肌、神经元、血管内皮和多种免疫细胞也有表达。此外ER-X富集于动物出生后类囊状微区,分子量为63 ku,在胎儿狒狒脑中分布较多,正常成年人含量基本为零,一般很难检查出来。有研究发现,当脑部缺血损伤时可被检测到,还需进一步研究其作用。雌激素受体具体分布位置,见Tab 1[3-5]。
2 雌激素受体信号通路及其甲基化
2.1 雌激素受体信号通路雌激素作为一种类固醇激素,可以直接通过质膜与细胞内的ERα和ERβ相互作用,通过与DNA序列结合来发挥直接作用,或者与GPER1和ERα和ERβ的相互作用来激活细胞内信号级联反应。由于细胞和分子事件的差异导致基因表达调节不同,其中雌激素受体复合物可以直接或间接地与DNA结合。雌激素介导的信号传导事件可以分为基因组和非基因组。基因组机制涉及基因转录,主由核 ER介导,以及在称为雌激素反应元件(EREs)的特定DNA序列上与染色质的直接相互作用[6]。非基因组作用模式由膜性受体介导,首先使细胞内的第二信使被激活,从而产生更多的反应,比如抗氧化和兴奋细胞等作用。
在中枢神经系统疾病中,雌激素受体对中枢神经系统的保护尤为关键。更年期雌激素水平下降可导致妇女出现情绪波动、认知功能的变化和记忆力减退。脑源性神经营养因子(BDNF)可以调节突触可塑性,当ERβ被激活时,小鼠脑中的BDNF蛋白显著增加,极大增强了脑部神经元分化能力,有效缓解记忆缺失症状。Broughton等[7]首次发现,在脑血管内皮细胞GPER-1也广泛表达,而且雄性小鼠脑GPER-1表达随着脑缺血程度增加而增加。种种迹象表明,雌激素受体在脑部神经系统疾病的发生发展进展中发挥重要作用。
2.2 雌激素受体甲基化雌激素甲基化过程中,在DNA甲基转移酶(DNA methyltransferases, DNMTs)的催化下,甲基从S-腺苷酸甲硫氨酸(SAM)转移到胞嘧啶残基的5-碳,从而在基因启动子胞嘧啶(C)-磷酸(p)-鸟嘌呤(CpG岛)[8]中形成5-甲基胞嘧啶(5mC)。CpG岛的表观遗传修饰改变了基因表达模式。当一个基因的启动子区域或转录结合位点甲基化程度变高时,它会抑制转录活性。因为雌激素受体本身不与任何DNMT酶结合。例如,ERα就会招募共同调节因子,如核受体相互作用蛋白(NRIP1);随后招募额外的辅抑制因子,如组蛋白去乙酰化酶(HDAC1)和多梳复合物2(PRC2)。组蛋白3(H3)和组蛋白4(H4)具有特别长尾,因此最常被修改。HDAC和zeste同源物2(EZH2)增强子(PRC2的酶促部分)首先使活化组蛋白标记脱乙酰,然后在启动子核小体中的组蛋白赖氨酸残基上放置抑制性甲基。EZH2和NurD在内的的PRC2复合物与DNA甲基转移酶复合物(DNMT3a、DNMT3b、DNMT3l)结合,然后DNMT使CpG岛甲基化[9-10]。总之,连接体ERα通过影响CpG岛中组蛋白和胞嘧啶残基的甲基化状态在基因转录调控中发挥作用。ERs在甲基化过程中招募共同抑制子(HDAC1、PRC2)和不同的蛋白质复合物(PRC2、NurD)。ERα、辅抑制因子和不同蛋白复合物之间的相互作用导致雌激素反应元件中CpG岛甲基化。仅激活DNMT是不够的,染色质重塑和组蛋白的甲基化也是阻止基因转录的关键。
3 雌激素受体甲基化与中枢神经系统疾病
雌激素和中枢神经系统疾病关系异常密切,本身雌激素具有一定的脂溶性,所以可以透过血脑屏障进入中枢神经系统,且雌激素也可以在脑部合成,通过重建突触,提高脑部血流量,促进神经再生。一般而言,雌激素受体发挥保护神经的作用主要通过扩张血管、增加脑部血流量、调节血管一氧化氮(nitric oxide, NO)水平、抑制动脉粥样硬化、拮抗兴奋性氨基酸(excitatory amino acids, EAA)毒性、抑制炎性因子表达、调控下丘脑-垂体-肾上腺轴、改善ERK信号通路、提高多巴胺等神经递质合成、减低PI3K/Akt 磷酸化、调节细胞内Ca2+水平、抑制β淀粉样变性,从而促进神经细胞增生,改善记忆和认知功能,营养保护神经系统。
3.1 雌激素受体甲基化与脑缺血卒中缺血性脑卒中是指由于脑部组织缺血缺氧导致部分区域神经功能发生改变。流行病学研究发现,绝经前妇女心脑血管疾病的发病率较低,而绝经后妇女与男子之间没有显著差异。相关实验表明,在成年啮齿动物皮层中,ERα启动子高度甲基化,但在神经元缺血性损伤后甲基化水平急剧而迅速地下降,这也解释了雌激素受体在脑缺血模型中对血管具有保护作用[11]。脑缺血/再灌注期间血管功能发生障碍,而NO是重要的血管舒张因子,影响血管的舒缩变化,雌激素通过结合其特异性受体,提高一氧化氮合酶基因表达[12],抑制血管平滑肌细胞的迁移和增殖来降低动脉硬化程度。缺血性脑卒中独立危险因素之一是同型半胱氨酸(homocysteine, Hcy)的改变,通过甲硫氨酸循环产生S-腺苷酸甲硫氨酸,从而改变DNA的甲基化状态,导致多种疾病的发生。刘松等[13]学者通过对狭窄大脑中动脉斑块雌激素受体ERα基因甲基化状态的研究发现,随着ERα甲基化和Hcy的升高,颅内不稳定斑块的形成加速。这也为脑血管病易感患者的筛检和识别提供依据。

Tab 1 Characteristics and distribution of estrogen receptor

Fig 2 Estrogen receptor methylation process
脑缺血时,脑内释放出大量的EAA,造成钙离子内流,氧自由基增多,从而对神经元造成损伤,而雌激素和谷氨酸受体大量共存细胞内,通过同一细胞受体,降低其自身受体甲基化水平来干扰EAA代谢,清除氧自由基,保护神经元。同时脑部缺血损伤也会导致促炎症因子比如IL-1β的上调,IL-1β通过激活β-连环蛋白途径对上皮间质转化(epithelial mesenchymal transformation, EMT)进行诱导,转录因子TWIST1的上调使得ERα基因启动子的甲基化水平下降,进而抑制胶质细胞的活化。细胞凋亡是脑缺血损伤后的重要表现,长期低氧会导致神经元内质网损伤,从而促使相关细胞凋亡。雌激素受体GPER激活,对内质网应激产生抑制,从而减少了神经元细胞的死亡,缓解脑缺血/再灌注损伤,其机制在韩子伟等[14]模拟脑缺血中GPER对内质网细胞凋亡研究中得以论证。有趣的是,ERβ基因多态性存在种族、地域上的差异,其不同基因酶切多态性使得脑缺血患者患病的易感性和年龄表达存在着不同的影响。这与ERα基因表达恰恰相反。两受体甲基化在脑缺血疾病中的表达差异有待进一步研究其机制。
3.2 雌激素受体甲基化与抑郁症抑郁症是一种高患病率、高自杀率及高复发率的脑部神经退行性疾病。围绝经期抑郁症高发于卵巢退化后的中老年人群,一些调查报告显示出雌激素受体水平的波动与围绝经期抑郁症的易感性密切相关。动物实验表明,老年雌性大鼠中当雌激素受体处于低甲基化状态时,可以调节外在刺激对机体的反应,增强了脑部神经的保护,一定程度上减少了抑郁行为的发生。其中ERαrs9340799 AA基因型和rs2234693 TT基因型已被证明与绝经后激素使用修饰的终生抑郁风险相关,而ER-βrs4986938 AA基因型也与重性抑郁症的患病率较高有关[15]。通常而言,抑郁症会导致HPA轴的糖皮质激素分泌增加,从而使促肾上腺皮质激素释放激素(corticotropin-releasing honnone, CRH)分泌过多,而雌激素可以通过其受体影响CRH水平,进而影响下丘脑-垂体-肾上腺轴,从而减轻焦虑和抑郁样行为发生。Birgit等[16]利用亚硫酸氢盐测序测定CRH基因中启动子特异性CpG甲基化。染色质免疫沉淀(ChIP)测定用于确定ERβ与CRH基因近端启动子区域的结合,随着ERβ甲基化程度增加,抑郁行为普遍增加。
细胞外调节蛋白激酶ERK信号通路是研究抑郁症发病机制的另一路径。雌激素通过激活BDNF受体TRK来激活丝裂原活化蛋白激酶(MAPK),从而活化ERKK1/2调节基因转录,发挥抗抑郁作用。宫谷谷等作者利用ERβ选择性激动剂二苯基丙腈来测试小鼠抑郁作用中BDNF的表达,发现低甲基化状态下ERβ增加了海马CA1中的PERK1/2表达,从而发挥抗抑郁作用[17]。神经递质假说在抑郁症的发病机制研究中占据很大一部分。其中5-羟色胺(5-hydroxytryptamine, 5-HT)是重要的中枢神经递质,参与HPG轴功能调节。雌激素受体激活可增加中缝背核中5-HT的合成,同时5-HT通过5-HT受体(5-HTA)的激活从而发挥其抗抑郁的作用。Kajta等[18]通过药物诱导小鼠模拟成人产生抑郁行为,通过气相色谱法和串联质谱法确定的产前给药在小鼠大脑中积累,导致整体雌激素受体的DNA低甲基化,并改变特定基因中甲基化DNA的水平。抑郁样行为的诱导和Htr1a/5-HT信号传导的损害,发现GPER1甲基化提高在药物诱导的抑郁样症状的传播中起关键作用。现代研究发现,动物海马体中炎症细胞的增加也会导致抑郁行为。一种名为NLRP3炎症小体出现在视野之中,它参与了因雌激素缺乏而导致的情感障碍类疾病。徐永军等[19]通过对炎症因子的研究,发现雌激素的缺乏,其受体高甲基化的表达会导致NLRP3炎症小体活化,从而导致海马体神经炎症以及抑郁和焦虑。这可能是以后治疗雌激素缺乏导致抑郁症疾病的潜在方向。
3.3 雌激素受体甲基化与阿尔茨海默病(AD)阿尔茨海默病(Alzheimer’s disease,AD)是指患者伴有认知功能减退、人格及行为障碍为特征,表现一类脑部神经系统病变疾病。其中β-淀粉样蛋白(Aβ)沉积,线粒体功能障碍和氧化应激反应,Tau蛋白异常以及Ca2+的流失是导致AD发病的主要原因。AD在绝经后妇女中更普遍。脑源性雌激素的消耗是 AD 发展的重要机制。脑萎缩是AD 主要的病理改变。雌激素受体的激活可以增强神经的可塑性,这与学习记忆的形成和改变密切相关。亚诺夫等[20]通过术后切除卵巢年轻和年老的Fischer大鼠,来检测海马CA1和CA3区Esr1表达和ERα启动子甲基化。研究发现,相对于CA3,CA1区Esr1表达降低与CA1区DNA甲基化增加有关,尤其是第一个CpG位点。此外,远端CpG位点11-17的差异甲基化与衰老期间或长期激素缺乏后Esr1表达的改变有关。
AD主要病理特征为过度磷酸化的Tau蛋白异常集聚形成的神经纤维缠结。更年期AD患者的大脑中雌激素缺乏。淀粉蛋白质(Abeta)和雌激素缺乏症都可以与tau激酶相互作用。实验表明,蛋白质磷酸酶2A(protein phosphatase 2A,PP2A)可能是ER表达的决定因素,雌激素受体的高甲基化导致PP2A(L309)去甲基化增加使AD大脑中PP2A活性降低,导致异常高磷酸化tau的去磷酸化功能受损。而ERα36基因沉默可以抑制ERK/AKT信号通路的激活,增加tau蛋白磷酸化。但Xiong等[21]在研究两种雌激素受体时,发现二者对AD患者中Tau蛋白磷酸化发挥了不同的作用,ERα增加miR-218的表达,以抑制其特异性靶标蛋白质酪氨酸磷酸酶α(protein tyrosine phosphatase α,PTPα)的蛋白质水平。PTPα的下调导致糖原合酶激酶-3β(导致活化)和蛋白质磷酸酶2A(导致失活)的异常酪氨酸高磷酸化。相反,ERβ通过限制miR-218水平和恢复miR-218水平,以此拮抗ERβ对tau磷酸化的衰减来抑制tau磷酸化。Ca2+信号在维持正常的脑功能中起着非常重要的作用,雌激素受体激动剂可有效加快神经元细胞内的Ca2+内流,加快了ERK 磷酸化,对神经起到保护作用,伴随着ERα的高甲基化会抑制Ca2+的内流。
淀粉样前体蛋白(APP)可以被α-、β-、γ-蛋白酶分解产生Aβ。而Aβ会对脑部神经产生很强的毒性,引发脑部神经元死亡病变。Tang等[22]通过在HEK293细胞中加入达全长的淀粉样前体蛋白(β-amyloid protein precursor,APP),发现低甲基化状态下的ERα对APP的合成过程起到了一定的抑制作用,且减弱了GSK3β活性。AD中ERβ的作用也不可忽视,在部分AD病人脑中发现其核ERβ数量显著下降。TIan等[23]发现,在老年大鼠AD模型中ERβ的低甲基化表达减少了海马体中的Aβ沉积,有效改善了其记忆下降问题。当然,既往的研究中氧化应激损伤和线粒体功能障碍也与AD有关。随着AD患者大脑中雌激素受体水平下降,线粒体氧化酶活性也同时降低,雌激素受体甲基化的高表达会导致线粒体功能障碍,脑部能量储备下降,诱发AD发病。
3.4 雌激素受体甲基化与帕金森病(PD)帕金森病(Parkinson’s disease,PD)是一种常见的神经系统变性疾病,最主要的病理改变是中脑黑质多巴胺(dopamine,DA)能神经元的变性死亡。雌激素受体ERα、ERβ和GPR30在纹状体和黑质中均已被证明存在,其中ERα是作用于神经保护的主体。陈荣杰等[24]利用聚合酶链反应-限制性片段长度多态性方法,对PD患者进行ERβ基因多态性分析,发现ERβ基因AluⅠ、RsaⅠ的酶切多态性不影响 PD 和 PDD 的患病风险。Kim等[25]。研究发现,雌激素受体ERβ低甲基化可以诱导RNF146的激活,从而对PAR聚合酶-1(PARP1)诱导的中风细胞死亡具有神经保护作用,可以预防由PD引起的细胞死亡。
炎症反应在PD中起着关键作用,药理学研究发现,当GPR30通过与雷诺昔芬结合时可发挥抑炎作用,从而抑制PD的慢性炎症进展。ER可以激活AKt信号转导机制,使其磷酸化,诱导Bcl-2等靶基因的表达。生育三烯酚(tocotrienols,T3s)是维生素E家族的成员,具有抗氧化特性,并且在PD等神经退行性疾病的发病机制中是起神经保护作用。雌激素受体ERβ作为γT3/δT3刺激后PI3K/Akt信号传导的上游介质。高度纯化的γT3/δT3直接在体外与ERβ结合,并且在SH-SY5Y细胞中敲除ERβ可消除γT3/δT3依赖性细胞保护和Akt磷酸化。这从侧面反映出ERβ甲基化表达水平的高低是直接影响T3治疗后早期PD发展的关键步骤。同时,一种噻二唑丙烯酰胺衍生物(XCT790)作为最具选择性ERα反向激动剂应用于PD的治疗。一般情况下,当ERα存在于自噬体中时,随着化合物的诱导自噬反应的增加,这种定位却并没有随之丧失,这说明ERα可以通过其自身动力学来调节这种反应。在相关模拟临床小鼠PD的模型中我们不难看出,一定条件下,自噬作用通过抑制ERα的活性,致使其甲基化水平提高。XCT790通过诱导自噬清除有毒蛋白聚集体,同时缓解运动协调障碍,并对黑质中的多巴胺能神经元发挥神经保护作用,消除了神经元细胞中α-突触核蛋白,这也为我们治疗PD提供了一个新的思路。
胰岛素样生长因子-1(insulin-like growth factor-1,IGF-1)是一种内源性肽,通过与雌激素受体相互作用,从而增强对大脑神经的保护。IGF-1可以改善MPTP诱导的运动缺陷,改善纹状体中DA及其代谢物含量的降低以及黑质(substantia nigra,SN)中TH-IR神经元的损失。实验表明[26],GPER的低甲基化激活可能有助于IGF-1的表达。从而对MPTP / MPP诱导的多巴胺能神经元损伤的神经保护作用。
3.5 雌激素受体甲基化与精神分裂症精神分裂症是一种由多组症状群组成的慢性疾病,临床表现各异。该病发病机制不明确,现代研究认为可能与海马代谢失调、前脉冲抑制(prepulse inhibition,PPI)等有关。通常女性发病较同龄男性晚,且在绝经期和产后期发病几率增加。精神分裂症的主要特征是大脑信息加工过载,同时感觉运动门控受损是导致该异常的重要机制之一。在利用苯丙胺诱导小鼠精神分裂症模型中,在基础条件下,使用 ERα或ERβ激动剂进行预处理可有效阻断苯丙胺诱导的PPI破坏。使用优先ERβ调节剂进行急性治疗可避免PPI[27]。说明雌激素受体低甲基化状态下可以增强PPI从而减缓病情发展。随着年龄的增加,其脑部海马区域的ERα甲基化表达水平下降。
有研究表明,ERα其中一个单核苷酸多态性(single nucleotide polymorphism,SNP)位点在精神分裂症患者中普遍存在,同时它与患者前额叶的ERα甲基化高低息息相关[28]。但是,在研究过程中发现多巴胺失调也会引起精神分裂症出现,在精神分裂症患者脑中海马神经元常常出现异常,这在雌激素缺乏患者中更为常见。当抗精神病药物和雌激素受体结合时往往效果更好。但许多患者对这些精神病药物容易上瘾,当临床反复用药时,多巴胺释放明显减少[29]。此类研究目前尚处于实验观察阶段,后续期待更多的发现。
4 结语
伴随着雌激素的衰退和身体机能的下降,脑部中枢神经系统疾病发病率不断提高,且雌激素主要通过其受体对中枢神经系统机制产生作用。表观遗传学是近年来兴起的学科,它从DNA基因角度揭示了相关脑部神经系统疾病的发病病因。雌激素受体甲基化对脑部疾病国内研究不甚很多,其作用机制尚不明确,还有许多问题和难点去研究,与相关疾病的研究才刚刚开始,尚需要对表观遗传修饰关系展开进一步的研究,以期揭示其具体作用机制。
猜你喜欢
家庭医学(下半月)(2020年3期)2020-05-30
天津医科大学学报(2019年6期)2019-08-13
分析化学(2017年12期)2017-12-25
家教世界·创新阅读(2017年7期)2017-08-09
奥秘(2016年6期)2016-07-30
分忧(2016年3期)2016-05-05
安徽医科大学学报(2015年9期)2015-12-16
现代检验医学杂志(2015年2期)2015-02-06
沈阳医学院学报(2014年4期)2014-12-27
遗传(2014年3期)2014-02-28
